
Remember me
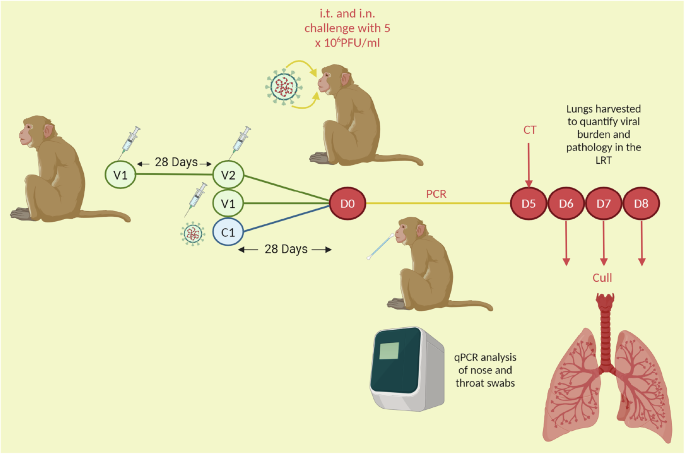
A cohort of ninety RhMs and CyMs either received a primary challenge, one or two doses of a vaccine candidate (an mRNA vaccine, a DNA vaccine, a formalin-inactivated vaccine (FIV) or the ChAdOx-vectored vaccine36,37,38,39,40, Table 1) or were in the control arm of the studies and were not immunised. Animals were subsequently challenged or re-challenged with a high viral dose of SARS-CoV-2 (5 ×106 PFU of Victoria/1/2020), delivered intratracheally and intranasally. There was a 28-day interval between the second/only vaccine dose and challenge in all but the FIV group that were challenged 14 days post-FIV vaccination. There was also a 28-day interval between the primary challenge and secondary challenge in the re-challenge cohort (Fig. 1). During acute infection, nasal and throat swabs were collected for qPCR. The lungs of each animal were harvested six-to-eight days post-challenge, for histopathology assessment, and qPCR of viral genomic RNA (gRNA) was performed on bronchoalveolar lavage (BAL) fluid to ascertain viral burden in the lung. The ChAdOx study and RhM versus CyM study included animals that were culled at earlier or later timepoints and these animals were excluded from the analysis of clinical and lung viral load outcomes (see Table 2 for group breakdown). Lung histopathology was scored using a scoring system defined by Salguero et al.36, which involved the scoring of seven lung pathology parameters from zero to four, with a score of four corresponding to more marked pathology, and a cumulative score of 28, across all parameters, describing maximum severity36.
Fig. 1: Schematic of pre-clinical study timeline.
The day of challenge post-immunisation is D0. The immunisation strategies varied. For two dose regimens, there was a 28 day interval between vaccine dose one (V1) and two (V2). There was a 28 day interval between second/only vaccine dose and challenge, in all but the FIV group, that were challenged 14 days post-FIV vaccination. There was also a 28 day interval between the primary challenge (C1) and secondary challenge (D0) in the re-challenged cohort. During acute infection, nasal and throat swabs were collected for qPCR. Animals were culled 6-8 days (D6-D8) post-challenge, and the lungs were harvested.
Table 1 Summary of vaccines included in studyTable 2 Number of macaques in each subgroup of study for each post-challenge outcome investigatedSerology data, including spike and receptor binding domain (RBD) binding antibody measured by a standard ELISA protocol, standardised seasonal human coronavirus (HCoV) antigen binding antibody measured by MSD, standardised Ig isotype and IgG subclass titres measured by a multiplex bead assay and neutralising antibody measured by micro neutralisation assay (MNA), was collected across all preclinical studies and the data is summarised in Supplementary Figs. 1, 2, 3 and 4, respectively. SARS-CoV-2-reactive T cell data measured by a standard IFNγ ELISpot protocol, and whole blood immunophenotyping data was also collected for each of the preclinical studies, except the re-challenge study. The ELISA, MNA, ELISpot and whole blood immunophenotyping was described and reported in the original pre-clinical publications36,37,38,39,40. In our study, data from these pre-clinical publications has been compiled along with pre-immunisation serology, spike antibody isotype and IgG subclass data to investigate SARS-CoV-2 CoPs.
The range of vaccine platforms, doses and regimens and the inclusion of a re-challenge cohort serves as an opportunity to compare pathology outcomes following SARS-CoV-2 challenge and identify shared CoPs (Fig. 2). Here, we focus on 4 possible outcomes to describe the protection against severe infection; LRT disease burden, evaluated by the 1) lung histopathology scores and 2) viral load in the lungs, and protection against URT infection measured by 3) nasal and 4) throat viral loads.
Fig. 2: SARS-CoV-2 challenge outcome analysis.
Box and whisker plots demonstrating the success of each immunisation strategy for a clinical protection determined using a histopathology scoring system, b limiting lung viral burden determined by BAL PCR 6-8 days post-challenge, c controlling throat viral load over the course of infection (area-under-the curve of the throat swab PCRs) (d) controlling nasal viral load over the course of infection (area-under-the curve of the nasal swab PCRs). Each datapoint represents an animal. Box plots show the group median and inter-quartile range (IQR), and whiskers connect the maximum and minimum values, extending no further than 1.5x IQR (data beyond whiskers are outliers). Kruskal Wallis test and Dunn’s multiple comparisons test performed. Two-tailed Spearman correlations between lung histopathology scores and immune responses with 95% confidence intervals (e) spike IgG, f neutralising antibody, g ELISpot. AUC area under the curve, P/C post-challenge.
It was found that two high doses of the mRNA vaccine and two doses of the DNA vaccine protected against severe infection as measured by the histopathology scores (Dunn’s multiple comparisons test, p = 0.0025 and p = 0.0074, Fig. 2a).
BAL qPCR 6-8 days post-challenge of macaques that received two high doses of the mRNA vaccine significantly differed to the unvaccinated control group (Dunn’s multiple comparisons test, p = 0.0025, Fig. 2b).
Interestingly, only immunity generated after natural infection was able to protect against URT infection as indicated by the nose and throat PCR area under the curve (AUC) data (Figs. 2c and 2d). Only re-challenged macaques had significantly lower viral AUCs to the unvaccinated control group (Dunn’s multiple comparisons test, p = 0.0246 and p = 0.0064, respectively, Figs. 2c and 2d). Re-challenge macaques also had significantly lower throat viral load AUCs than the macaques that received two low dose mRNA vaccines (Dunn’s multiple comparisons test, p = 0.0019, Figs. 2c and 2d).
SARS-CoV-2 specific humoral and T cell responses have been associated with the outcome of vaccine pre-clinical studies, clinical trials and vaccine follow-up studies2,15,40,41,42,43,44,45,46,47,48,49,50. To identify shared immunological signatures associated with protection, we performed integrated analysis across vaccine platforms. We first performed correlation analysis using immunogenicity data collated from the vaccine and challenge trials discussed above. We observed a significant positive correlation between histopathology scores and spike IgG (Fig. 2e; Spearman correlation, r = −0.5662, p < 0.0001), receptor-binding domain (RBD)-binding IgG (Supplementary Fig. 5; Spearman correlation r = −0.5059, p = 0.0001) and NAbs (Fig. 2f; Spearman correlation, r = −0.775, p < 0.0001), but an insignificant relationship between histopathology scores and IFNγ ELISpot (Fig. 2g; r = −0.2201, p = 0.1415).
Vaccine outcome parameters of interest included histopathology score, lung viral load 6-8 days post-infection, throat viral load AUC during infection and nasal viral load AUC during infection. To support ML predictions, each of these outcomes were made binary; either protected or pathology, and either high or low virus. Each macaque was assigned to a binary category based on defined cut-offs (histopathology score = 6.75, lung viral load AUC = 120,000, throat viral load AUC = 590,000 and nasal viral load AUC = 1.5 ×107), for each outcome parameter (Supplementary Fig. 6–9). Critically, depending on the outcome parameter used to define vaccine efficacy in pre-clinical models, the immunisation strategies varied in success. An immunisation strategy that minimised pathology post-challenge did not necessarily mediate viral control. Only 19 out of a total of 69 macaques (27.5%), which had data on each of the four post-challenge outcomes, had agreeable virology outcomes and clinical outcomes. Clinical outcomes were the most agreeable with the BAL PCR 6-8 days post-challenge outcome, with 49 of 69 macaques having high lung virus plus high pathology or low lung virus plus low pathology (71%). There was also a strong significant positive correlation between lung viral load and histopathology scores (Supplementary Fig. 10a; Spearman correlation, r = 0.4782, p < 0.001). However, histopathology scores did not correlate strongly with throat viral load (Supplementary Fig. 10b, Spearman correlation, r = 0.1233, p = 0.3096) and correlated weakly with nasal viral load (Supplementary Fig. 10c, Spearman correlation, r = 0.2394, p = 0.0444). Therefore, clinical protection (ie. protection from pathology) and protection from high viral load, particularly in the upper respiratory tract, may need to be considered separately.
Immunological signatures that differentiate mild from more severe pathology outcomes post-SARS-CoV-2 challengeMacaques do not experience severe COVID-19 symptoms or succumb to infection, therefore in this study, clinical outcomes were stratified by histopathology scores that were assigned to macaques following histopathological assessment of respiratory organs 6-8 days post-SARS-CoV-2 challenge. The histopathology scoring system is described in Salguero et al.36. Based on these scores the macaques were classified into protected or pathology groups (cut-off = 6.75, Supplementary Fig. 6).
In-life computed tomography (CT) imaging scores day 5 post-challenge was used to define clinical severity in animals that were not assigned a histopathology score (n = 6, from the control group in the re-challenge study), or were culled 13-14 days post-challenge (n = 4 from ChAd-vectored vaccinated group and n = 4 from the ChAd-vectored control group), because at this timepoint, the infection would have resolved and the histopathology score would not accurately reflect acute pathology. Animals that were not culled between the timeframe of 6-8 days post-SARS-CoV-2 challenge, and did not have a CT scan performed during acute infection, were excluded from this analysis (n = 12, see Table 2 for group breakdown). CT scores correlated with histopathology score (Pearson correlation, r = 0.624 and p < 0.0001) and there was strong agreement between protection or pathology group assignment when histopathology or CT score were used (71%, and of the 29% with conflicting categorisation, 50% of macaques had scores around the cut-off ( ± 1.5)). The ChAdOx study and re-challenge control macaques, for which we relied on CT scores alone for clinical outcome classification, had CT scores that were far from the median CT score cut-off, therefore we were confident in the placement of these macaques into protected and pathology groups based on their CT score. Correlation filtering (corr) was not performed as a) immune features commonly correlate and b) all immunological measurements pre-challenge were included in the dataset, and so inter-timepoint correlations would exist (see Supplementary Fig. 11).
Using a supervised ML platform, SIMON, 171 machine learning algorithms were tested and 26 successful models were identified based on model selection criteria of having an area under receiver operator curve (AUROC) for the training set that is greater than 0.7 and a test set AUROC greater than the training AUROC. The best performing model was built using the Boosted Classification Trees (ada) algorithm (train AUROC 0.8771 and test AUROC 0.9074, Supplementary Fig. 12). Of the 158 immune features provided, 21 immune features scored a variable of importance score above 50, and so best predict clinical protection when modelled with the ada algorithm (Fig. 3b, Supplementary Fig. 13).
Fig. 3: SARS-CoV-2 challenge clinical outcome analysis.
a Venn diagram showing the agreeance, or lack thereof, between each outcome post-challenge. b Variables of importance for clinical protection – predictors of protection in blue and predictors of pathology in red. c Polar plot showing the difference in magnitude of the humoral responses between the protected and pathology groups. The abundance of (d) baseline CD4+ T cells phenotyped by flow cytometry predicting protection and e CD14hiCD16hi monocytes two weeks post-V2 predicting pathology. Datapoints represent each animal, and the bars show group means with standard deviation. Two-sided Mann Whitney test performed.
SARS-CoV-2 neutralising antibody (NAb) titres best modelled protection from SARS-CoV-2 challenge, as the highest variable importance score (VIS) was recorded for this feature (VIS 100, Figs. 3b and 3c) and high NAb titres differentiated ‘protected’ from ‘pathology’ outcomes with significance (p < 0.0001, Supplementary Fig. 13). High SARS-CoV-2 spike binding IgG titres were also strong predictors of protection. High SARS-CoV-2 spike IgG binding titres on the day of challenge measured by three different serological assays were variables of importance (VOI) and significantly differed between the ‘protected’ and ‘pathology’ groups; spike-conjugated bead assay (VIS 99, p < 0.0001), ELISA (VIS 89, p < 0.0001), and MSD (VIS 86, p < 0.0001, Supplementary Fig. 13), validating its prediction potential, even when measured by different assays. A high RBD IgG response on the day of challenge was also predictive of clinical protection (MSD; VIS 79, p < 0.0001, ELISA; VIS 63, p = 0.0002, Supplementary Fig. 13). High SARS-CoV-1 spike IgG (VIS 70, p < 0.0001, Supplementary Fig. 13) also predicted outcome upon challenge.
As early as two weeks post-vaccine dose one (V1), high spike IgG and NAb titres predicted protection (spike IgG by ELISA VIS 65, and NAbs by MNA VIS 63) and there was a significant difference in spike IgG (p = 0.0184) and NAb (p = 0.0032) titres between the pathology and protected groups from this timepoint (Supplementary Fig. 13). High spike-binding and neutralising antibody titres from week four post-V1 (S IgG; VIS 69, p = 0.003, MNA; VIS 66, p = 0.0004, Supplementary Fig. 13) also contributed to the modelling of protection and significantly differentiated protected from pathology groups. Similarly, from week two post-V2, high spike IgG titres and RBD IgG titres predicted clinical protection (spike IgG ELISA; VIS 66, p < 0.0001, RBD IgG ELISA; VIS 65, p < 0.0001, Supplementary Fig. 13). Early serological predictors of clinical protection were investigated further to ensure this result was not influenced by outliers, which was not the case (see matched timecourse graphs, Supplementary Figs. 14a–c).
IgG subclasses also emerged as predictors of protection by SIMON analysis. IgG subclass titre and its ratio with respect to the other subclasses, was incorporated into the one parameter for SIMON analysis (Supplementary Fig. 15). Protected macaques fell into IgG4 and IgG3 subclass categories with higher titres and higher ratios (Fig. 3c), which significantly differed from the IgG subclass category distribution of macaques in the pathology group (Chi-square test); IgG4 (VIS 68, p = 0.0029, Supplementary Fig. 13) and IgG3 (VIS 63, p = 0.0026, Supplementary Fig. 13). Protected macaques fell into IgG2 and IgG1 high titre categories (Fig. 3c), which significantly differed from the IgG subclass distribution of macaques in the pathology group (Chi-square test); IgG2 (VIS 59, p = 0.0011, Supplementary Fig. 13) and IgG1 (VIS 55, p = 0.0025, Supplementary Fig. 13). High IgM titre was also a predictor (VIS 61) and titres significantly differed between the protected and pathology groups (p = 0.0001, Supplementary Fig. 13).
The ada algorithm also modelled protection using cellular responses. An early T cell response was important in establishing a protection-associated immune profile as a high abundance of CD4+ (VIS 56, Fig. 3d) and CD4+CD95+ (VIS 64, Supplementary Fig. 13) T cells at baseline were predictors of protection and significantly differed between protected and pathology groups (p = 0.015 and p = 0.0265, respectively). A high frequency of CD14hiCD16hi cells at two weeks post-V2, was the only parameter predictive of pathology (VIS 64) and significantly differentiated pathology from protected groups (p < 0.0001) (Fig. 3e).
No singular immunisation regimen contributed to the trend of immune divergence that determined predictability (Supplementary Fig. 14d–f), therefore the predictors identified are not vaccine-specific. Clinical outcome could not be modelled using MNA titres alone, or spike IgG titres alone, with the ada algorithm. However, MNA and spike IgG titres together successfully modelled clinical protection with improved model performance (train AUROC of 0.9146 and test AUROC of 0.9444, data not shown). This highlights the potential for enhanced predictive power when a number of predictive immune parameters are used together to predict post-challenge outcomes.
Immunological signatures that differentiate low from high viral loads in the lung post-cull following SARS-CoV-2 infectionA major benefit of using animal models for the study of SARS-CoV-2 CoPs is that we can monitor the extent of viral control following a known exposure event with a known titre of virus.
qPCR on BAL fluid to measure SARS-CoV-2 viral RNA burden in the lung has been used by several groups as a protective endpoint in pre-clinical evaluation of candidate SARS-CoV-2 vaccines42,51. The frequency of macaques having both high lung viral load and pathology is 71% (Fig. 3a) and there was a significant correlation (r = 0.4782, p < 0.0001, Spearman Correlation, Supplementary Fig. 10a) between these outcomes. The relationship between virology and pathology in the lung, may therefore mean that LRT viral load burden can be used as a surrogate for severity of pathology. Therefore, in lieu of a veterinary pathologist or a robust histopathology scoring system, BAL PCR results can be reflective of clinical protection as was applied in mRNA-1273 pre-clinical trials42,51. We next look to determine if the immune parameters that predicted pathology also predicted viral control in the lung.
Of the 90 macaques, 69 had BAL qPCR data from 6-8 days post-challenge available (see Table 2 for group breakdown). Immune features, measured at all timepoints pre-challenge, were used in the attempt to model lung viral burden. SIMON used 174 machine learning algorithms and found 10 algorithms that successfully modelled viral titres in the lung. The best performing model was built using the LogitBoost algorithm (train AUROC 0.7847 and test AUROC 0.8214, Supplementary Fig. 12) and 20 of the 158 immune features were identified as predictors of lung viral load outcomes (Fig. 4a).
Fig. 4: Immune predictors of lung viral load post-SARS-CoV-2 challenge.
a Variables of importance for lung viral load –predictors of low viral load in blue. b Polar plot showing the difference in magnitude of the humoral responses between the low and high lung viral load groups. c Spike IgM on the day pre-challenge predicts low lung virus (d) SARS-CoV-2 reactive PBMCs two weeks post-V1 predict low lung virus. Datapoints in bar charts represent each animal, and bars show group means with standard deviation. Two-sided Mann Whitney test performed.
Many of the same parameters that predicted clinical protection, also predicted low lung virus (Fig. 4b). High SARS-CoV-2 spike IgM on the day of challenge was identified as the best predictor of low lung virus and titres significantly differed between macaques with high and low lung viral load (VIS 100, p < 0.0001, Fig. 4c, Supplementary Fig. 16). High spike IgG on the day of challenge was also predictive of low lung viral load and titres significantly differed between the low and high lung viral load groups (Bead; VIS 96, p < 0.0001, ELISA; VIS 89, p < 0.0001 and MSD; VIS 80, p = 0.0004, Fig. 4b, Supplementary Fig. 16). High RBD IgG titres, measured by ELISA and MSD on the day of challenge predicted lung viral burden (ELISA; VIS 85, p < 0.0001 and MSD; VIS 69, p = 0.0019, Fig. 4b, Supplementary Fig. 16), as did high NAb titres (VIS 77, p < 0.0001, Fig. 4b, Supplementary Fig. 16). Similar to the predictors of clinical protection, high spike IgG responses as early as two weeks post-V1 could predict low viral load in the lung post-challenge (VIS 63, p = 0.0235), as could high spike IgG titres four weeks post-V1 (VIS 68, p = 0.0057) and two weeks post-V2 (VIS 55, p = 0.0039). High RBD IgG titres from four weeks post-V1 (VIS 56, p = 0.0043) and two weeks post-V2 (VIS 72, p = 0.0001) were also identified as early predictors of low lung viral load post-challenge (Supplementary Fig. 16). The identification of early serology markers as lung viral load predictors was investigated further to ensure this result was not influenced by outliers, which was not the case (see timecourse graphs, Supplementary Figs. 17a and b). No singular immunisation regimen contributed to the trend of immune divergence that determined predictability (Supplementary Fig. 17c–e). Lung viral load outcome could not be modelled using spike IgM titres alone, spike IgG titres alone or MNA titres alone with the LogitBoost algorithm.
The protective humoral response on the day of challenge was also reactive to other beta CoV spike proteins; SARS-CoV-1 (VIS 82, p = 0.0006, Fig. 4b, Supplementary Fig. 16), HCoV-OC43 (VIS 59, p = 0.0036, Supplementary Fig. 16) and MERS-CoV (VIS 51, p = 0.0513, Supplementary Fig. 16). As per the predictors of clinical protection, IgG subclass also emerged as a predictor of low lung viral load. Higher titres and ratios of IgG4 (VIS 91, p = 0.0023), IgG3 (VIS 78, p = 0.0028) and IgG2 (VIS 67, p = 0.0129) were associated with low lung viral load (Chi-square test, Fig. 4b, Supplementary Fig. 16). High IgG1 titres were also associated with low lung viral load (VIS 79, p = 0.0036, Chi-square test, Fig. 4b, Supplementary Fig. 16).
We did not find CD14hiCD16hi monocytes or early T cell subset frequencies to be predictive of lung viral load. Instead, we observed SARS-CoV-2 reactive PBMCs, measured by IFNγ ELISpot two weeks post-V1, to positively predict low lung virus and significantly differentiate macaques that went on to have high or low lung viral burden (VIS 55, p = 0.0329, Fig. 4d).
Immunological signatures that differentiate low from high viral loads in the upper respiratory tract over the course of acute SARS-CoV-2 infectionViral control in the upper respiratory tract and the immune factors facilitating this, may also be critical in understanding the endpoint of reduced SARS-CoV-2 transmission. Limiting viral transmission, as a vaccination endpoint secondary to clinical protection from severe disease, was less of a focus in the midst of the pandemic, as it was difficult to ascertain at the clinical trial phase because of the SARS-CoV-2 control measures in place at the time. Whilst the macaque studies reported here did not measure transmission directly in a horizontal transmission setting, lower viral titres in the nose and throat in the 6-8 days post-challenge would be indicative of reduced transmission risk, based on the increased likelihood of transmission via air droplets from sneezing and coughing were viral load at these mucosal sites high.
It is also important to note the poor correlation between histopathology scores and throat viral titres (r = 0.123, p = 0.3096, Spearman Correlation, Supplementary Fig. 10b), and histopathology scores and the nasal viral titres (r = 0.2394, p = 0.0444, Spearman Correlation, Supplementary Fig. 10c), signifying the lack of agreement between these endpoints. The overlap between the virology and clinical outcomes, i.e. the overlap between the macaques in low URT virus and protected groups, or high URT virus and pathology groups, was also unconvincing, 59.7% for throat virus and 54.5% for nose virus (Fig. 3a). This supports the treatment of clinical and URT viral load as distinct outcomes.
Models were successfully built for throat virus control only. All 90 macaques had throat swabs collected over the course of infection and AUC was calculated using the available throat swab PCR data. Of 174 machine learning algorithms, 6 models of viral titres in the throat were successfully built. The best performing model was built using the Boosted Classification Trees (ada) algorithm (train AUROC 0.7684 and test AUROC 0.7901, Supplementary Fig. 12) which identified 23 immune features with a VIS greater than 50 (Fig. 5a).
Fig. 5: Protection against high viral burden in the upper respiratory tract.
a Variables of importance for throat viral control – predictors of low viral load in blue and predictors of high viral load in red. b Polar plot showing the difference in magnitude of the humoral responses between the low and high viral load groups. c Polar plot showing the difference in magnitude of the cellular frequency between the low and high viral load groups. d Venn diagram showing the overlap of variables of importance for each of the modelled outcomes.
Humoral responses on the day of challenge predicted low throat viral load over the course of acute SARS-CoV-2 infection in macaques (Fig. 5b). High spike IgG on the day of challenge, measured by three different assays, were predictors of low throat viral loads (MSD; VIS 100, p < 0.0001, ELISA; VIS 91, p = 0.0002, Bead Assay; VIS 64, p = 0.0053, Fig. 5b and Supplementary Fig. 18) as were high RBD-binding IgG titres on the day of challenge (ELISA; VIS 79, p = 0.0002, MSD; VIS 78, p = 0.0004, Fig. 5b and Supplementary Fig. 18). High NAb titres on the day of challenge also predicted throat viral control and significantly differentiated low from high viral loads (VIS 65, p = 0.0017, Fig. 5b and Supplementary Fig. 18). The humoral response that supports throat viral control was also reactive to SARS-CoV-1 spike protein (Fig. 5b and Supplementary Fig. 18; VIS 81, p = 0.0003). High spike-binding IgM on the day of challenge also predicted low throat viral loads post-challenge (Fig. 5b and Supplementary Fig. 18; VIS 79, p = 0.0023).
From two weeks post-V1, a high spike-binding antibody titre was predictive of low throat viral load (ELISA; VIS 60, p = 0.0116, Supplementary Fig. 18). Antibody titres at later timepoints post-vaccination had higher variable importance scores and so are of greater importance in the modelling of throat viral load outcomes. For example, spike-binding IgG, measured by ELISA, four weeks post-V1 had a VIS of 88 and two weeks post-V2 had a VIS of 68, and both significantly differentiated low from high throat viral load groups (p < 0.0001 and p = 0.0005, respectively, Supplementary Fig. 18). RBD-binding IgG four weeks post-V1 had a VIS of 51 and significantly differentiated low from high throat viral load groups (p = 0.0004, Supplementary Fig. 18). To ensure these serological parameters were not identified as predictors of throat viral load because of outliers, time-course plots were generated (see Supplementary Fig. 19a and b). No singular immunisation regimen contributed to the trend of immune divergence that determined predictability (Supplementary Fig. 19c–e). Throat viral load outcome could not be modelled using spike IgG titres alone or MNA titres alone with the ada algorithm.
Higher pre-immunisation (study baseline) IgG responses to HKU1 spike and SARS-CoV-2 nucleocapsid were also predictive of high viral load in the throat over the course of infection; HKU1 spike IgG (MSD; VIS 86, p = 0.0159, Supplementary Fig. 18), SARS-CoV-2 nucleocapsid IgG (MSD; VIS 67, p = 0.0328, Supplementary Fig. 18). A high frequency of CD14hiCD16hi intermediate monocytes and granulocytes two weeks post-V1 also predicted high throat viral load (both VIS 59). A high abundance of CD14hiCD16hi intermediate monocytes in peripheral blood significantly differentiated high from low throat viral loads (p = 0.006, Fig. 5c and Supplementary Fig. 18).
The abundance of several different T cell subsets throughout the vaccination regimen predicted low throat viral load (Fig. 5c). A higher abundance of CD4+CD95+ T cells (VIS 66, p = 0.0103, Fig. 5c and Supplementary Fig. 18), CD8+PD1+ T cells (VIS 62, p = 0.0172, Fig. 5c and Supplementary Fig. 18), CD4+PD1+ T cells (VIS 55, p = 0.0397, Fig. 5c and Supplementary Fig. 18), CD4+ T cells (VIS 51, p = 0.0283, Fig. 5c and Supplementary Fig. 18) and CD3+ T cells (VIS 51, p = 0.0279, Fig. 5c and Supplementary Fig.
Comments (0)