Study design and sample collection
This retrospective study aimed to explore HDAC3 as a potential immunotherapy biomarker in aNSCLC through a multi-phase approach. In the discovery phase (n = 22), proteomic analysis was used to identify differential AAbs between responders and non-responders to ICIs, revealing 127 AAbs—51 upregulated and 76 downregulated in non-responders. Considering the pivotal role of PD-L1 as a biomarker in lung cancer immunotherapy, an additional cohort (n = 22) with available PD-L1 expression data was analyzed to evaluate the independent predictive value of AAbs. This plasma cohort also underwent targeted protein microarray analysis.
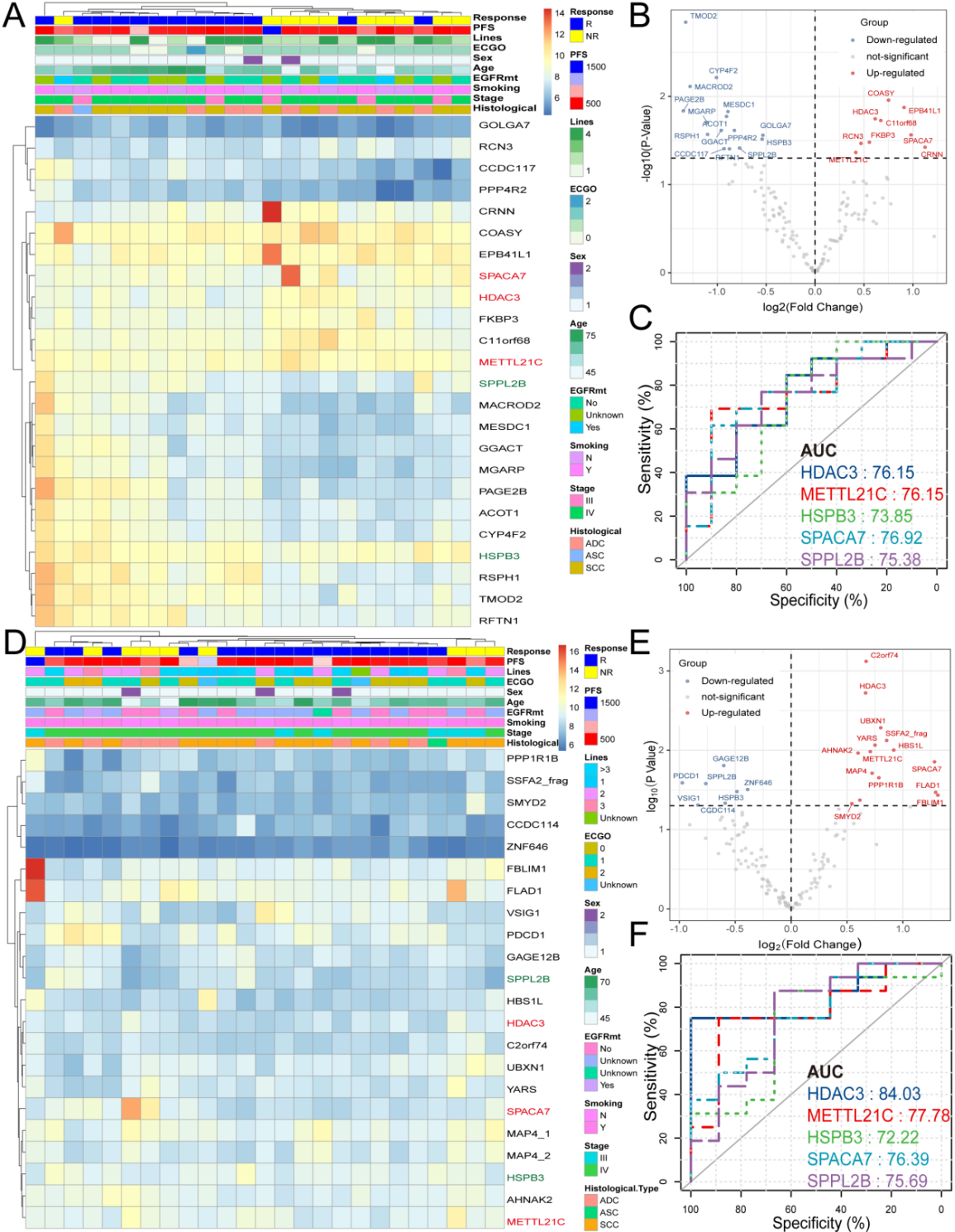
Patients for both the discovery phase and validation phase were randomly selected to ensure unbiased sample representation. To ensure methodological rigor, the discovery cohort (n = 22) and validation cohort (n = 34) were designed with matched pre-treatment and post-treatment pairs. A balanced distribution of samples was maintained, with pretreatment samples (discovery: 22 vs. validation: 23) and post-treatment samples (discovery: 23 vs. validation: 25) allocated at a near 1:1 ratio. This is further supported by the absence of significant differences in clinical characteristics between cohorts, including gender distribution, ECOG, histology, and disease stage (P > 0.05). In the validation phase, we conducted aNSCLC-focused microarray analysis, comparing AAb levels in responders and non-responders at two time points. We identified 24 and 22 prognostic AAbs (p < 0.05), including five key AAbs (HDAC3, METTL21C, HSPB3, SPACA7, SPPL2B) with consistent expression patterns. We also performed a dynamic assessment in a cohort of patients at baseline and subsequent time points to track AAb trends, constructing a risk score model that predicted PFS. Additionally, HDAC3 expression was validated in 21 FFPE samples via immunohistochemistry and was associated with higher TGFβ1, PD-L1 infiltration and lower CD8+ T cells infiltration, while single-cell RNA sequencing assessed gene expression differences between HDAC3+ and HDAC3− cells, revealing significant pathway enrichments. Finally, we utilized si-HDAC3 and a C57BL/6 mouse model to evaluate the role of HDAC3 in lung cancer cells, the therapeutic effects of RGFP966 (an HDAC3 inhibitor) and anti–PD-1 antibodies on tumor growth and immune cell infiltration. This comprehensive approach aims to elucidate the role of HDAC3 in immune modulation and its potential as a biomarker for enhancing aNSCLC immunotherapy outcomes. Detailed clinical characteristics were shown in Tables S1 and S2.
Between 2016 and 2022, a total of 138 plasma samples were collected from 78 aNSCLC patients, including 67 pre-treatment samples (T0), 47 post-treatment samples (T1) within three months of ICIs therapy, 24 post-treatment samples (T2) at the time of three months. Additionally, 42 formalin-fixed paraffin-embedded (FFPE) samples were obtained from 42 aNSCLC patients who received ICI monotherapy (Nivolumab, Pembrolizumab, Sintilimab, or Tirellizumab) at the Cancer Hospital, Chinese Academy of Medical Sciences. All plasma samples were collected using ethylenediaminetetraacetic acid, centrifuged at 3000 rpm at 4 °C for 10 min, and stored in 2 ml conical tubes at -80 °C until the microarray assay. FFPE samples were stored at room temperature.
The inclusion criteria for patients were: (1) biopsy-confirmed aNSCLC with stage III or IV disease and with clinical data (gender, age, smoking history, ECOG, histological type, staging) and follow-up data (efficacy evaluation at 3 months and PFS); (2) treatment with ICI monotherapy; and (3) ICIs used as either first-line or later-line therapy. Patients were excluded if they met any of the following conditions: (1) a concurrent diagnosis of another cancer; (2) non-primary lung tumors; (3) concomitant autoimmune diseases; or (4) use of other immunosuppressive agents (e.g., steroid medication). The efficacy of immunotherapy was assessed using the Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1. Patients were categorized as “responders” if they achieved complete remission, partial remission, or stable disease within three months of ICI treatment, and as “non-responders” if they experienced disease progression. This study was approved by the Ethics Committee of the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College (No. 19–019/1804). All procedures were conducted in accordance with the principles of the Declaration of Helsinki.
HuProt™ microarray and aNSCLC-focused microarray
Standard experimental procedures for high-density microarrays have been described in our previous studies [19, 20]. Briefly, the HuProt™ microarray (CDI Labs, Mayaguez, PR) consisted of approximately 21,000 proteins. The microarray was retrieved from − 80 °C storage and blocked with bovine serum albumin (BSA, 9048-46-8, Sigma) for 1.5 h. Following the blocking step, the microarray was incubated with plasma samples at a 1:1000 dilution for 1 h. After washing with 0.1% phosphate-buffered saline-Tween 20, the plates were incubated with Alexa Fluor 647 goat anti-human IgG (109-605-003, Jackson) diluted in 5% BSA. The microarray was then scanned using a GenePix 4300 A microarray scanner (141095, Molecular Devices) with a 635 nm excitation laser. Signal intensities of IgG for each protein were quantified using GenePix Pro v.6.0 software (Molecular Devices). For the validation phase, candidate AAbs identified during the discovery phase were selected and printed onto 2 × 7 sub-arrays to create an aNSCLC-focused microarray. The experimental procedures for the aNSCLC-focused microarray were similar to those used for the high-density microarray, except for the blocking and dilution buffer, which was 3% BSA.
Immunohistochemistry validation
All patient samples underwent hematoxylin-eosin staining and were meticulously reviewed by two experienced pathologists to identify cancer lesions. Immunohistochemistry (IHC) was performed on FFPE samples following dewaxing and heat-induced antigen retrieval. After washing, the samples were treated with a 3% hydrogen peroxide solution to quench endogenous peroxidase activity. Primary antibodies, including rabbit anti-human IgG antibodies specific to histone deacetylase 3 (HDAC3, ab32369, Abcam), were applied at a 1:500 dilution following blocking with rabbit serum. After incubation, the samples were treated with a 1:200 dilution of HRP-labeled goat anti-rabbit IgG secondary antibody (GB23303, Servicebio) for 50 min at room temperature. Diaminobenzidine (DAB) was used for color development, and the nuclei were counterstained with hematoxylin (G1004, Servicebio). The results were examined under a white light microscope (E100, Nikon) and quantified using CaseViewer 2.4 (3DHISTECH, Hungary) software. Protein expression levels were quantified using an H-score, which was calculated based on the intensity of the stain and the percentage of positive tumor cells, with scores ranging from 0 to 300. Staining intensity was categorized as negative (0), weak (1), moderate (2), or strong (3), and the percentage of positive cells was scored from 0 to 100. The H-score was determined by multiplying the intensity score by the percentage of positive cells. An H-score below 60 was classified as low expression, while an H-score of 60 or above was considered high expression.
Single-cell and bulk-RNA sequencing analysis of HDAC3
Data from GSE131907 (n = 21), which includes NSCLC tumor samples (including advanced stage lung tumors, lymph node metastases, and brain metastases), as well as data from GSE182434 (n = 5), consisting of four tumor samples from DLBCL and one tonsil sample from a patient with tonsillitis, were retrieved. Clinical data and metadata were obtained from the original study [21, 22]. Additionally, immunotherapy-related dataset GSE218989 [23] (n = 339 NSCLC) was annotated and utilized for immunotherapy prediction analysis. Raw data underwent rigorous quality control using the “Affy” package [24] within R software, involving the computation of average values for multiple probes corresponding to a single gene. The risk score was calculated using the formula: risk score = Σ (Expression * Coefficient) and generated through the use of the “ggrisk” package.
Initially, quality control was performed on the GSE131907 data using “Seurat” [25] package, which focused on single-cell RNA-seq data analysis, particularly for clustering and dimensionality reduction. Using Seurat, we were able to perform quality control, normalization, and integration of datasets, followed by clustering cells based on gene expression profiles. Quality control was focusing on cell quality by filtering based on the number of detected genes (minimum: 300, maximum: 6000), mitochondrial gene percentage (0–15%), hemoglobin gene percentage (0–0.1%), and ribosomal gene percentage (minimum: 1–100%). Genes expressed in fewer than three cells were excluded. Malignant epithelial cells were then isolated. Based on the expression status of HDAC3, these malignant epithelial cells were classified into ‘HDAC3-positive’ and ‘HDAC3-negative’ groups. Copy number variations (CNVs) analysis was subsequently conducted on these cells using the “inferCNV” [26] package, which was applied to detect CNVs from single-cell RNA-seq data, calculating CNV scores for each cell type. By analyzing the expression levels of genes across cells, inferCNV allowed us to infer CNVs that could distinguish cancerous from non-cancerous cells, providing insights into the genomic landscape of the tumors. Additionally, “GSEABase” [27] package was used to perform rank-based Gene Set Enrichment Analysis (GSEA). It provided the tools to access, organize, and work with gene sets, allowing us to evaluate whether specific pathways or biological processes were significantly enriched in our data, helping to interpret the functional implications of gene expression changes in the context of cancer.
To investigate the differences in interactions between HDAC3+ and HDAC3− tumor cells and immune cells (including exhausted Tfh, exhausted CD8+ T cells, cytotoxic CD8+ T cells, CD8+/CD4+ mixed Tfh, CD8+ low T cells, CD4+ Tfh, NK cells, naive CD8+ T cells, naive CD4+ T cells, and microglia / macrophage cells), the “CellChat” [28, 29] package was employed to analyze cell-cell communication. It facilitated the identification of signaling pathways involved in tumor-immune cell interactions. By constructing cell-cell communication networks, we could explore the potential roles of specific ligand-receptor pairs in mediating immune responses in the tumor microenvironment. Specific categories within the CellChat database, such as “Secreted Signaling,” “ECM-Receptor,” and “Cell-Cell Contact,” were examined, with a minimum cell count criterion of three applied.
Multiple Immunofluorescence
FFPE tissue sections of 4–5 μm thickness were prepared, followed by dewaxing and rehydration. Antigen retrieval was performed, and endogenous peroxidase activity was blocked using an antibody blocking solution. Sequential immunostaining was carried out for each target antigen, including primary antibodies against rabbit anti-human IgG for HDAC3 (ab32369, 1:250, Abcam), CD8A (ab237709, 1:500, Abcam), TGFB1 (ab215715, 1:500, Abcam), PD-L1 (#13684S, 1:800, CST), and mouse anti-human pan cytokeratin IgG (ab7753, 1:250, Abcam). This was followed by incubation with secondary antibodies: HRP-labeled goat anti-rabbit IgG (GB23303, 1:500, Servicebio) for HDAC3, CD8A, and TGFB1 and HRP-labeled goat anti-mouse IgG (GB23301, 1:500, Servicebio) for pan cytokeratin. Tyramide signal amplification (TSA) was employed, followed by microwave treatment to remove the TSA-antibody complex, allowing for subsequent rounds of antibody labeling. After immunostaining, cell nuclei were counterstained with 4’,6-diamidino-2-phenylindole, and slides were coverslipped for scanning. Microscopy (ECLIPSE C1, Nikon) and scanning (Pannoramic MIDI, 3DHISTECH) were used for result interpretation. The CaseViewer 2.4 (3DHISTECH) and ImageJ software were utilized to quantify the number and percentage of positive cells. All results were independently reviewed by two experienced pathologists.
Cell and animal experimentCell culture and transfection
Human non-small cell lung cancer cell lines A549 and H1299 were obtained from the American Type Culture Collection (ATCC). Cells were cultured in RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin/streptomycin (Gibco, USA) at 37 °C in a humidified atmosphere containing 5% CO₂. Cells were passaged when they reached 70–80% confluence. The Lewis (catalog KG070) mouse lung cancer cell line was cultured in DMEM supplemented with 10% fetal bovine serum at 37 °C and 5% carbon dioxide. All experiments were conducted using mycoplasma-free cells.
RNA interference and lentiviral infection
To construct the RNA interference (RNAi) vector targeting HDAC3, single-stranded DNA oligonucleotides with the interference sequences were synthesized. These oligonucleotides were annealed to form double-stranded DNA and ligated into the pre-digested RNAi vector. The ligation products were transformed into competent bacteria, and correct clones were confirmed by sequencing.
Lentiviral vectors expressing HDAC3 shRNA were generated by co-transfecting HEK-293T cells with the RNAi plasmid and packaging plasmids using Lipofectamine 2000 (Invitrogen, USA). After 48 h, the culture supernatant was collected, filtered, and concentrated to obtain the lentivirus. A549 and H1299 cells were infected with the concentrated lentivirus for HDAC3 knockdown. Stable clones were selected by incubation with puromycin (1 µg/mL, Sigma-Aldrich, USA) for 2 weeks. Successful knockdown of HDAC3 was confirmed by qPCR and Western blot analysis.
For qPCR, specific primers for the target genes (HDAC3 and GAPDH) were designed using Primer3 software based on the gene sequences obtained from the GenBank database. The primers used for qPCR are as follows: HDAC3_Forward (CGCTATGTGGATACGCTGCTTTA), HDAC3_Reverse (GCAACCAGGATTTAT ACAAGGAGGA), GAPDH_Forward (GTCTCCTCTGACTTCAACAGCG), and GAPDH_Reverse (ACCACCCTGTTGCTGTAGCCAA). For Western blot, The primary antibodies used were Anti-β-Actin (1:1000, CST, #4970S) and Anti-HDAC3 (1:5000, Abcam, ab32369).
Cell proliferation assays (CCK-8)
Cell proliferation was assessed using the CCK-8 assay (Dojindo, Japan). A549 and H1299 cells were seeded in 96-well plates and allowed to adhere overnight. Cells were transfected with HDAC3-targeting shRNA or control vectors as described above. At 24, 48, and 72 h post-transfection, CCK-8 reagent (10 µL) was added to each well, and the cells were incubated for 2 h at 37 °C. Absorbance was measured at 450 nm using a microplate reader. The proliferation rate was calculated relative to the control group.
Colony formation assay
For the colony formation assay, cells in the logarithmic growth phase were trypsinized with 0.25% trypsin and dissociated into a single-cell suspension by pipetting. The cell suspension was resuspended in complete culture medium. The cell suspension was then serially diluted and seeded in 6-well plates at a density of approximately 300 cells per well. Cells were evenly distributed and allowed to settle for 2–3 weeks in the incubator, with medium replacement every 4–5 days. Once visible colonies appeared, culture was terminated. The medium was discarded, and cells were carefully washed twice with PBS. Cells were then fixed with 1 mL of 4% paraformaldehyde for 15 min, followed by staining with crystal violet solution for 20 min. The staining solution was gently washed off with deionized water, and the colonies were allowed to air dry. Colonies were counted and photographed.
Cell cycle analysis
Cell cycle distribution was analyzed by flow cytometry. A549 and H1299 cells were transfected with HDAC3-targeting shRNA or control vectors, and at 48 h post-transfection, cells were harvested, washed with cold PBS, and fixed in 70% ethanol at 4 °C overnight. Fixed cells were then stained with propidium iodide (PI) solution containing RNase A for 30 min at 37 °C.
Animal experiment
RGFP966 is a specific inhibitor of HDAC3, N-(o-aminophenyl)carboxamide, purchased from MedChemExpress (catalog HY-13909) [30, 31]. Male C57BL/6 mice (6–8 weeks old) were sourced from Shanghai GemPharmatech. These mice were housed in a specific pathogen-free animal facility with free access to food and water. They were maintained at 22 °C ± 2 °C with a relative humidity of 70%, under a 12-hour light/dark cycle. To establish the NSCLC model, Lewis (1 × 10^7) cells were injected into the flanks of C57BL/6 mice. Tumors were monitored and measured with calipers every 2–3 days. After 8 days of Lewis cell implantation, tumor volumes reached approximately 100 mm³. C57BL/6 mice carrying Lewis cells were randomly divided into three groups (n = 3), including a control group, an anti-PD-1 mAb group, and an RGFP966 plus mAb anti-PD-1 group. RGFP966 (catalog HY-N0504) was dissolved in PBS and administered to mice at a dosage of 25 mg/kg daily. The PD-1 monoclonal antibody (catalog BE0146, BioXCell) was dissolved in PBS and intravenously administered at a dose of 200 µg three times a week. The control group received only intravenous PBS. Treatment continued until tumor volume exceeds 3000 mm3. Mice were euthanized with an overdose of 0.5% pentobarbital sodium, and tumors were excised, photographed, and weighed. The resected tumors were subjected to flow cytometry analysis.
Statistical analysis
All statistical analyses were conducted using IBM SPSS Statistics 24 (Chicago,
Illinois, USA), R version 4.2.1, Sanger plot (http://www.sangerbox.com), and Hiplot (https://hiplot.com.cn/home/index.html). The Mann-Whitney U test was used for group comparisons. Sensitivity, specificity, and ROC curves were calculated using the “pROC” [32] and “ROCR” [33] packages in R. The “maxstat” [34] package in R was employed to determine the optimal cutoff values for categorizing high- and low-expression groups for survival analysis in both the discovery and validation phases. A p-value of < 0.05 (two-tailed) was considered statistically significant for all analyses.

Comments (0)