
Remember me

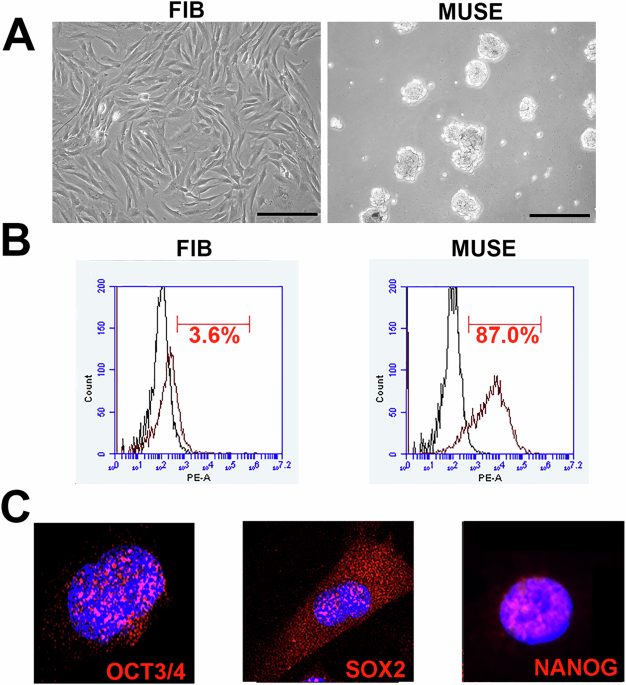
The iPSC lines used in this study were generated from skin biopsies as described in [32] obtained at the Neuro Center, Neurology, Kuopio University Hospital. All biopsy donors gave written informed consent. The research in human subjects is performed in accordance with the ethical standards of the Declaration of Helsinki and approved by the Research Ethics Committee of the Northern Savo Hospital District, Kuopio, Finland with the ethical permit 16/2013. Studies on FTD patient-derived iPSC-neurons are performed with permission 123/2016 from the Research Ethics Committee of the Northern Savo Hospital District. In this study, iPSCs were generated and characterized from six Finnish FTD patients (53–77 years) clinically diagnosed with bvFTD and three age-matched healthy control individuals according to [33]. The FTD patient iPSCs were derived from three C9-HRE carriers and three sporadic (non-genetic) FTD patients. In addition to those lines, a commercially available line from an FTD patient carrying the C9-HRE was used in some experiments. The line was purchased from EBiSC (cell line name: UCLi001-A, Biosamples ID: SAMEA3174431). EBiSC bank acknowledges University College London and UCL Queen Square Institute of Neurology with support from the NIHR UCLH Biomedical Research Centre, EFPIA companies and the European Union (IMI-JU’). Repeat-primed PCR (AmplideX® PCR/CE C9orf72 Kit, Asuragen) was performed on genomic DNA extracted from corresponding blood samples and iPSCs to confirm the presence or absence of the C9orf72 HRE in the carriers (C9+,1 line > 60 repeats; 2 lines >145) and sporadic FTD (C9−, <30 repeats) and healthy controls (<30 repeats). Patient data were pseudonymized and handled using code numbers. All the iPSC lines were tested negative for mycoplasma regularly (Lonza, LT07-418). iPSCs were maintained on Matrigel (Corning, 356230) coated 3.5 cm dishes (Sarstedt) in E8 medium (Essential 8™ Medium; Gibco,1517001), supplemented with, 1×E8 supplement (Gibco, A1517001) and 0.5% (v/v) penicillin/streptomycin (Pen/Strep; Gibco,15140122) at +37 °C and 5% CO2. Cell culture medium was replaced every second day. Once confluency of 60–80% was reached, iPSCs were split by incubating for 3 min at +37 °C in EDTA (Invitrogen,15575020) containing E8 medium and the whole medium was changed on the following day. For freezing, iPSCs were collected in E8 medium supplemented with 10% (v/v) heat inactivated FBS (Gibco,10500) and 10% (v/v) DMSO (Sigma D2650) and stored long-term in liquid nitrogen. After thawing the iPSCs were kept in E8 medium containing 5 µM Y-27632 2HCl (Selleckchem, S1049) for 24 h.
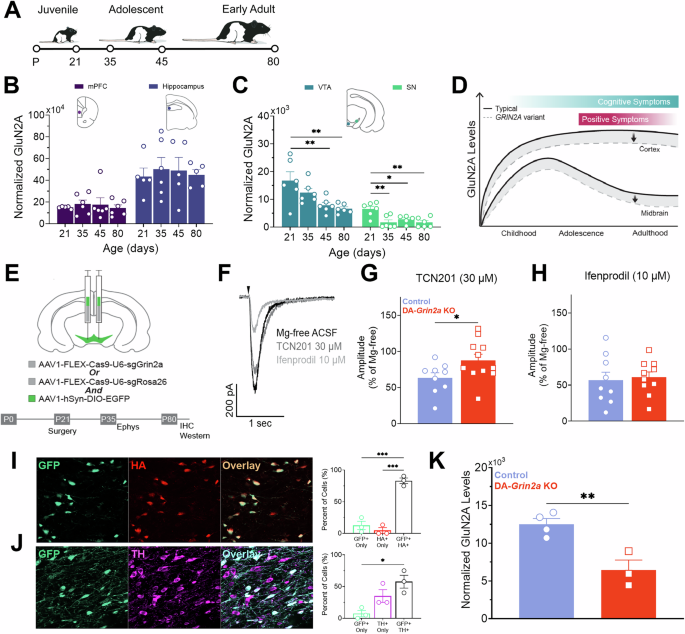
Differentiation of iPSCs to neuronal progenitor cells and cortical neuronsTo induce neuronal differentiation, the iPSCs were seeded onto Matrigel-coated 6-well plates in E8 medium. When the colonies reached appropriate size (~50% confluency), the medium was changed to E8 medium containing the two dual SMAD inhibitors: 10 µM SB431542 (GF-βRI inhibitor, Selleckchem, S1067) and 200 nM LDN193189 2HCl (BMP type I receptor inhibitor, Selleckchem, S7507). The concentration of the dual SMAD inhibitors was kept the same for the whole differentiation process and they were added to the medium every day. On day 1, to start neuronal differentiation, the whole medium was changed containing two parts E8 medium and one part neural differentiation medium (NDM) containing 1:1 DMEM F12/Neurobasal medium supplemented with 1% B27 (Thermo Fisher, 12587010) 0.5% N2 (Thermo Fisher, 17502048), 2 mM Glutamax (Thermo Fischer, 35050038), and 0.5% (v/v) Pen/Strep. On day two, a full medium change was performed containing two parts NDM medium and one part E8 medium. Starting from day three, the whole medium containing only NDM and the dual SMAD inhibitors was changed. When neuronal rosettes containing differentiated neuroepithelial cells appeared after approximately day 10–12, the medium was changed to NDM containing 25 ng/ml bFGF (100-18B, Peprotech) for about two days, depending on the rosette size and appearance. On day 14, the colonies containing the neuronal rosettes were manually scraped into small clusters and transferred onto ultra-low culture dish (ULA; 4615, Corning) with neuronal sphere medium (NSM) containing 1:1 DMEM F12/Neurobasal medium supplemented with 0.5% N2, 2 mM Glutamax and 0.5% (v/v) Pen/Strep. On the next day, the whole medium was carefully changed without disturbing the neuronal spheres and from that on, about ¼ of the medium was changed three times a week with NSM supplemented with bFGF. The spheres were manually dissected into smaller spheres to maintain a progenitor-state neural cell population and expanded at least once every 2 weeks. The spheres were cultured for up to eight months. The first experiments were performed after 30 days in culture, but most experiments were performed after 180+ days in culture. The spheres were dissociated with Accutase (Thermo Fischer, A1110501) and plated in NSM medium w/o bFGF onto plates double-coated with 0.1 mg/ml poly-L-ornithine hydrobromide (Sigma, P3655) in Dulbecco’s Phosphate-Buffered Saline (DPBS) overnight at +37 °C and 30 µg/ml laminin (Sigma-Aldrich, L2020) in DPBS for four hours (with density 1 × 106 cells/6-well plate or 100,000 cells/24-well plate) to allow acquirement of neuronal morphology. The neurons were maintained for 14 days in NSM medium before performing experiments. Medium samples were collected before fixing the neurons.
Immunocytochemistry, fluorescence in situ hybridization, and quantification of sense RNA fociFor immunocytochemistry (ICC), the neurons were fixed with 4% PFA for 20 min at RT and permeabilized in 0.25% Triton X-100 for 60 min at RT and washed twice with DPBS. The cells were blocked in 1% bovine serum albumin (BSA; Sigma) in DPBS for 60 min at RT and incubated with primary antibodies against MAP2 (1:100, Sigma, 9942), GFAP (1:400, Dako, Z0334), vGlut1 (1:300, Sigma, V0389), C9orf72 (1:400, Genetex, GT1553), MAP2 conjugated to CoraLite®594 Fluorescent Dye (1:400,Proteintech, CL594-17490), p62/SQSTM1 (1:200,Santa Cruz, sc-28359), TDP-43 conjugated to CoraLite® Plus 488 Fluorescent Dye (1:100,Proteintech, CL488-80002), or anti-phospho-histone γH2A.X Ser139 (1:400, Sigma, 05-636) overnight at +4 °C. The cells were then incubated with fluorescently labelled secondary antibodies Alexa Fluor 647, Alexa Fluor 594, or Alexa Fluor 488 (Invitrogen) for 1 h at RT. Afterwards, the coverslips were washed with DPBS and mounted with Vectashield Vibrance Antifade mounting medium containing 4, 6-diamidino-2-phenylindole (DAPI, Vector Laboratories) and imaged with Zeiss Axio Observer inverted microscope equipped with a Zeiss LSM 800 confocal module (Carl Zeiss Microimaging GmbH, Jena, Germany) or Olympus BX-51 fluorescent microscope with 40X or 60X objective. To analyze the neuronal and astrocyte composition of the cultures, five random regions in five ICC images stained with MAP2 and GFAP were analyzed and the percentage of these cell types was calculated.
For fluorescence in situ hybridization (FISH), diethyl pyrocarbonate DPBS (DEPC-DPBS) and DEPC-treated H2O were used. FISH was performed in an RNase-free environment and fluorescently labelled locked nucleic acid (LNA) TYE™ 563-(CCCCGG)3 probe (C4G2; Exiqon) was used to detect the sense foci. TYE™ 563-(CAG)6 (CAG; Exiqon) was used as a negative control probe. The cells on coverslips were permeabilized in 0.2% Triton X-100 for 15 min at RT and washed three times in DEPC-DPBS. The coverslips were handled through an ethanol series (70, 70, and 100%, 1 min each) followed by drying the coverslips for 10 min at RT. Next, pre-hybridization was performed in hybridization buffer containing 10% dextran sulfate (Millipore, 3730-100 ML), 10 mM Ribonucleoside Vanadyl Complex (NEB # S1402S - 200 mM), formamide (Midsci, IB72020), 20 × SSC (Ambion, AM9763), and 1 M sodium phosphate buffer, pH 7, in DEPC-H2O, at +66 °C for 30 min. The probes were denatured at +85 °C for 1 min 15 s and added to hybridization buffer at a concentration of 40 nM C4G2 or CAG probe and incubated with the coverslips at +55 °C for 4 h. Then, after washing in washing buffer 1 (0.1% Tween in 2 × SSC) for 5 min at RT and washing buffer 2 (0.2 × SSC) for 3 × 10 min at +60 °C, the coverslips were mounted with Vectashield Vibrance Antifade mounting medium containing DAPI and imaged with Zeiss LSM800 Airyscan confocal microscope. To quantify the sense RNA foci in the neurons from the three C9-HRE-carrying lines, ten to 18 regions per image and a total of 1767 neurons were analyzed. Of these, 297 neurons displayed sense RNA foci.
Neurofilament light chain Simoa measurementsNeurofilament light chain (NfL) levels in the culture medium were quantified with the Quanterix single molecule array (Simoa, Billerica, MA, USA) HD-X analyzer, using the Simoa® NF-light™ V2 Advantage kit (104073) according to the manufacturer’s instructions. Briefly, frozen medium samples were slowly thawed on ice, mixed, and centrifuged at 10,000 × g for 5 min at RT, and loaded to a 96-well plate. Samples were measured in duplicate and using 1:4 dilution. The Lower Limit of Detection of NfL was reported at 0.038 pg/ml and the Lower Limit of Quantification at 0.174 pg/ml.
Dipeptide repeat protein measurementsiPSC neurons were cultured for 14 days, then medium was collected and the cells snap frozen. The Mesoscale DPR immunoassay was performed as described in [34]. Briefly, the frozen cell pellets were thawed on ice for 10 min and lysed in RIPA buffer. DPR immune assay was performed on the MSD platform using streptavidin plates (MSD Gold 96-well Streptavidin SECTOR: #L15SA). The plates were coated overnight with biotinylated capture antibody in blocking buffer (PBS with 0.05% Tween-20, 1% BSA) at +4 °C. After washing (DPBS with 0.05% Tween-20), the plates were incubated for 2 h at RT on a shaking platform (140 rpm). After washing, sulfo-tag-labeled detection antibody diluted in blocking buffer was added and the plates were incubated for 2 h at RT on a shaking platform (140 rpm), followed by washing. The electrochemiluminescent signal was measured using a MESO QuickPlex SG120 instrument. Antibodies used for poly-GP measurement were biotinylated 18H8 (0.13 mg/ml) and sulfo-tagged 3F9 (1 mg/ml). Antibodies for poly-GA measurement were biotinylated 1A12 (1 mg/ml) and sulfo-tagged 1A12 (1 mg/ml). Total protein levels in all lysates were quantified using PierceTM BCA Protein Assay Reagents (Thermo Fisher: #23222 and #23224). After the incubation step for the poly-GP immunoassay, a fraction of the lysed sample was used to measure total protein concentration. The DPR antibodies were a kind gift from Prof. Dr. Dieter Edbauer.
3D culture and dendritic spine analysisFor dendritic spine analysis, the 10-mm microwell in a 3.5-cm glass-bottom microwell dishes (MatTek, P35G-1,5-10-C) were coated with 0.1 mg/ml poly-L-ornithine hydrobromide for two hours at +37 °C. Then, the coating was removed, and the dish left to dry overnight in the laminar flow hood at RT. Neuronal spheres were collected and dissociated using Accutase (~500,000 cells/0.5 ml) and mixed with green fluorescent protein (GFP) adeno-associated virus with multiplicity of infection (MOI) 5000. The GFP construct was packaged in AAV9 virus particles at the BioCenter Finland National Virus Vector Laboratory, University of Eastern Finland, Kuopio, Finland. Matrigel and cell/virus suspension was mixed in a ratio of 1:8 and a 100-µl drop was added to the coated microwells and incubated at +37 °C and 5% CO2 for at least 24 h to harden the Matrigel. Then NSM medium was carefully added. Half of the medium was changed three times per week. The cultures were cultured for additional up to 180 days for dendritic spine analysis. Dendritic spines from GFP-positive neurons were imaged with LSM 800 Airyscan confocal microscope. Serial Z-stacks of optical sections from dendritic segments were analyzed by NeuronStudio software (Computational Neurobiology and Imaging Center Mount Sinai School of Medicine, New York, Version 0.9.92 64-bit). Settings were adjusted as following: Volume: pixel dimensions were set to X = 0.066 μm (pixel width), Y = 0.066 μm (pixel height), and Z = 0.390 μm (voxel depth); Dendrite detection: attach ratio was set to 1.5, minimum length to 3 μm, discretization ratio to 1, and realign junctions to yes; Spine detection: maximum height of spines was set to 5.0 μm and minimum height to 0.2 μm, maximum width to 3 μm, maximum stubby size to 10 voxels, and minimum non-stubby size to 5 voxels; Spine classifier: neck ratio (headneck ratio) was set 1.1, thin ratio to 2.5, and mushroom size to 0.35 μm. To obtain a clear image, the image filter Blur-MP was run before the analysis and the dendritic spines were sub-grouped according to their morphology to mushroom, stubby, or thin spines according to NeuronStudio’s automatic settings using the Rayburst algorithm.
Calcium imagingiPSC neurons were loaded with calcium-sensitive fluorescent Fluo-4 dye (1×, Direct Calcium Assay Kit, Invitrogen, USA) in humidified incubator (5% CO2, +37 °C) for 30–40 min following by washout for 10 min at +37 °C and then for 5 min at RT in basic solution (BS) containing 152 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 10 mM glucose, 2 mM CaCl2, 1 mM MgCl2, pH adjusted to 7.4. Then, coverslips with the loaded cells were transferred to the recording chamber with constant BS perfusion (flow rate 2.5 ml/min, RT). Fluo-4 loaded cells were imaged using monochromatic light source (TILL Photonics GmbH) with an excitation light wavelength of 495 nm (exposure time 100 ms) and registered emission intensity at ≥520 nm. Fluorescence was visualized with 10X objective in Olympus IX-70 microscope equipped with CCD camera (SensiCam, PCO imaging, Kelheim, Germany) and recorded with TILL Photonics Live Acquisition at 1-Hz sampling frequency (1 frame per second). Setup was equipped with Rapid Solution Changer RSC-200 (BioLogic Science Instruments, Grenoble, France) allowing local application of various solutions, with fast exchange between them (~30 ms). The cells were sequentially treated with short (2 s) applications (separated by 2 min BS washout) of 100 μM GABA (Sigma), 100 μM glutamate (together with 30 μM NMDA receptor co-agonist glycine; both from Sigma), 50 mM KCl (depolarizing agent, to differentiate neuronal cells from glia), and 10 μM ionomycin (membrane-permeable calcium ionophore, to define the cells’ viability; Tocris).
Analyses of C9orf72 protein levels, TDP-43 cytoplasm-to-nucleus ratio, and p62/SQSTM1 vesiclesiPSC neurons were stained with antibodies against MAP2, C9orf72, and with DAPI, as described above. C9orf72 images were acquired using Zeiss LSM800 Airyscan confocal microscope. For the C9orf72 analysis, binary MAP2 masks were created by applying the Li thresholding method to the MAP2 images. Particles smaller than 2 µm were removed and holes within the cell bodies of a size between 0–4 μm2 were filled. To capture C9orf72 signal from the full cytoplasm, nuclear masking was applied. DAPI images were used to create nuclear masks by using the Otsu thresholding method, followed by removal of particles with a radius smaller than 5 µm. The C9orf72 signal was quantified as sum intensities of the fluorescence signal originating from the fluorescently labeled secondary antibody within MAP2-positive and DAPI-positive areas (defining the neuronal boundaries) per image. Sum intensities were normalized to MAP2-positive area per image. Images were processed using Fiji (2.3.0/1.53f51; [35]). Neurons were stained with MAP2 and TDP-43 or p62/SQSTM1 antibodies and DAPI, as described above. TDP-43 images were acquired using Zeiss LSM800 Airyscan confocal microscope. p62/SQSTM1 images were acquired using Olympus BX-51 fluorescent microscope. Images were processed using Fiji (2.3.0/1.53f51; [35]). For both analyses, MAP2 images were used to outline neurons and measure the total cell body area per image of neurons only. For TDP-43 analysis, MAP2 images were processed by enhancing the local contrast (blocksize = 127, histogram = 256, maximum = 3.10). Next, Otsu thresholding method was applied, followed by removal of particles with a radius smaller than 5 µm. To fill holes within cell bodies, particles of a size between 0–4 μm2 were included. After watershed segmentation, regions of interest were outlined without further area or shape exclusion. Sum intensity values of depicted area from TDP-43 images were acquired. Then, DAPI images were analyzed including only the nuclei of MAP2-positive cells. DAPI images were used to indicate nuclei and measure nuclear areas per image (for TDP-43 analysis). To segment nuclei, thresholding (Renyi’s entropy method) was applied, followed by the fill holes command. Finally, sum intensity values of DAPI-positive areas were extracted from TDP-43 images. TDP-43 signals were quantified as sum intensities of the fluorescence signal originating from the fluorescently labeled antibody targeted against TDP-43 within nuclear (DAPI-positive) and cytosolic areas (nuclear signal subtracted from signal within whole cell body [MAP2-positive areas]) respectively for each image. Sum intensities were normalized to nuclear and cytosolic area sizes respectively for each image. Cytosolic to nuclear TDP-43 signal ratio was calculated for each image of each cell line.
For p62/SQSTM1 image analysis, MAP2 images were converted into binary images using the Li thresholding method, and further processed by removing small objects (≤5 µm radius) and using watershed segmentation. Next, p62/SQSTM1-positive vesicles were analyzed from MAP2-positive regions by converting images into binary images using thresholding (Renyi’s entropy method). Only particles with a minimum area of 0.01–1 µm2 in size were included in the analysis. Number of p62/SQSTM1-positive vesicles per image was normalized to total MAP2 area of the same image. Average size and integrated density of p62/SQSTM1-positive vesicles per image and cell line were extracted from Fiji.
Nuclear morphology and DNA damage analysisiPSC neurons were stained with γH2A.X antibody and DAPI as described above. Nuclear masking used in γH2A.X analysis was created in Fiji based on the DAPI staining by applying unsharp mask (radius = 1, mask = 0.60), followed by enhancing local contrast (CLAHE) (blocksize = 127, histogram = 256, maximum = 3) and adding a median filter (radius = 3). After converting the image to binary, particles with a radius smaller than 5 µm were removed. Total nuclear area per image was acquired by measuring masked regions with an area of 1.5 µm2 or higher. For γH2A.X masking, any signal in the γH2A.X channel outside of the DAPI mask was removed. The remaining signal was measured as sum intensity of the fluorescence signal from the fluorescently labeled secondary antibody, and the sum intensity from one image was normalized to the total nuclear area in the image. To acquire γH2A.X-positive particle number and size, the remaining γH2A.X signal was transformed into binary by auto thresholding (MaxEntropy algorithm), followed by analyzing the particles with an area higher than 0.00001 µm2. Particle number per image was normalized to the total nuclear area in the image, and particle size was measured as the mean particle area (µm2) per image.
For the analysis of nuclear morphology, the neurons were stained with DAPI as described above and imaged with Zeiss LSM800 Airyscan confocal microscope. To segment the nuclei, the Fiji plugin StarDist [36] was utilized. From StarDist, the Versatile (fluorescent nuclei) built-in model was used, with the non-maximum suppression postprocessing parameters of 0.5 (Probability/Score threshold) and 0.6 (Overlap threshold). The acquired nuclear regions-of-interest were copied to the non-segmented image, and morphological parameters of each DAPI-positive particle were measured. The following morphological parameter was calculated per DAPI-positive particle: eccentricity [sqrt(a2-b2)/a], where a = major axis length and b = minor axis length.
The images were divided into two most similar groups based on the mean area of the DAPI-positive particles per image using K-means clustering. Python version 3.11.8 and the following libraries were used for clustering [37]. The data from each group was reshaped with the NumPy (version 1.26.2) module “reshape” using numpy.reshape(−1,1) prior to clustering. Data points from each group separately were clustered into two groups using the KMeans algorithm from the scikit-learn [38], (version 1.4.1) module “sklearn.cluster”.
To study the DNA damage response, iPSC-derived neurons were treated with 10 µM topotecan (Topotecan hydrochloride, Tocris, 4562) or DMSO vehicle for 1 h. The cells were fixed with 4% PFA and stained with conjugated MAP2 and γH2A.X antibody as described above. Images were processed using Fiji (2.3.0/1.53f51). The nuclear masking used for γH2A.X analysis was based on the DAPI staining. It was created by applying unsharp mask (radius = 1, mask = 0.60), followed by enhancing local contrast (CLAHE) (blocksize = 127, histogram = 256, maximum = 3) and a median filter (radius = 3). The image was converted to binary and particles with a radius smaller than 5 µm were removed. The total nuclear area per image was quantified by measuring masked regions with an area of 1.5 µm2 or higher. To mask the γH2A.X, only signals within the DAPI area were used. The remaining signal was measured as sum intensity of the fluorescence signal from the fluorescently labeled secondary antibody, and the sum intensity from one image was normalized to the total nuclear area in the image. To acquire γH2A.X-positive particle number and size, the remaining γH2A.X signal was transformed into binary by auto thresholding (MaxEntropy algorithm), followed by analyzing the particles with an area higher than 0.00001 µm2. Particle number per image was normalized to the total nuclear area in the image, and particle size was measured as the mean particle area (µm2) per image.
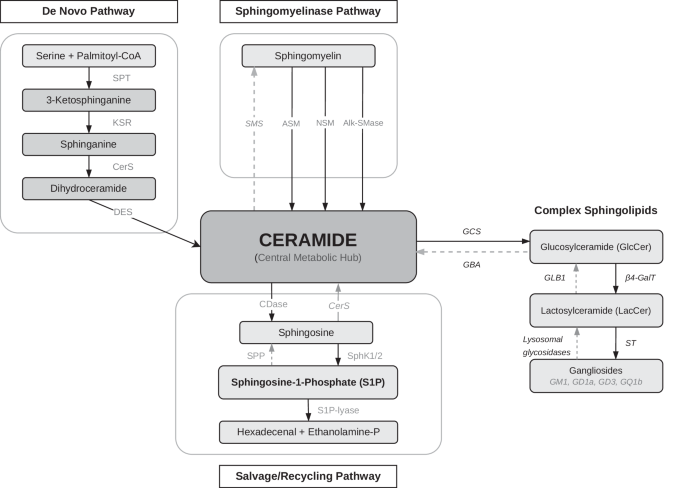
RNA sequencing and data processingRNA was extracted using the Direct-zol RNA Miniprep Plus (Zymo, R2072, USA, California). Bulk RNA sequencing (RNA-seq) was performed using RNA extracts from iPSCs and iPSC neurons. Library preparation and RNA sequencing was conducted by Novogene (UK) Company Limited. In brief, mRNA enrichment was performed with oligo(dT) bead pulldown, from where pulldown material was subjected to fragmentation, followed by reverse transcription, second strand synthesis, A-tailing, and sequencing adaptor ligation. The final amplified and size selected library comprised of 250–300 base pair (bp) insert cDNA and paired-end 150 bp sequencing was executed with an Illumina high-throughput sequencing platform. Sequencing yielded 22.3–27.1 million sequenced fragments per sample.
The 150-nucleotide pair-end RNA-seq reads were quality-controlled using FastQC (version 0.11.7) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were then trimmed with Trimmomatic (version 0.39) [39] to remove Illumina sequencing adapters and poor quality read ends, using as essential settings: ILLUMINACLIP:2:30:10:2:true, SLIDINGWINDOW:4:10, LEADING:3, TRAILING:3, MINLEN:50. Reads aligning to mitochondrial DNA or ribosomal RNA, phiX174 genome, or composed of a single nucleotide, were removed using STAR (version 2.7.9a) [40]. The remaining reads were aligned to the Gencode human transcriptome version 38 (for genome version hg38) using STAR (version 2.7.9a) [40] with essential non-default settings: –seedSearchStartLmax 12, –alignSJoverhangMin 15, –outFilterMultimapNmax 100, –outFilterMismatchNmax 33, –outFilterMatchNminOverLread 0, –outFilterScoreMinOverLread 0.3, and –outFilterType BySJout. The unstranded, uniquely mapping, gene-wise counts for primary alignments produced by STAR were collected in R (version 4.1.0) using Rsubread::featureCounts (version 2.8.2) [41], ranging from 17.2 to 20.7 million per sample. Differentially expressed genes (DEGs) between experimental groups were identified in R (version 4.2.0) using DESeq2 (version 1.36.0) [42] by employing Wald statistic and lfcShrink for FC shrinkage (type = “apeglm”) [43], and correcting for sequencing batch. Pathway enrichment analysis was performed on the gene lists ranked by the pairwise DEG test log2FCs in R using clusterProfiler::GSEA (version 4.4.4) [44] with Molecular Signatures Database gene sets (MSigDB, version 2022.1) [45].
Data processing, visualization, and statistical analysesTo test whether data points within the experimental groups were normally distributed, Shapiro-Wilk test was used. To test for significance between two different experimental groups, two-tailed independent samples t-test was used for normally distributed data and Mann Whitney U test for not normally distributed data. To test for significance between more than two groups, one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test or Sidak´s multiple comparison test was used if data points were normally distributed. Otherwise, Kruskal-Wallis test followed by Dunn’s multiple comparisons test was used. For grouped variables, two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was used. To assess statistical independence between categorical variables, the Chi-Square test was used. All tests were performed using GraphPad Prism software (version 8.4.3 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com). To check for and remove potential outliers in the data, GraphPad Prism’s Outlier Identification (ROUT method, Q = 1%) was used. The data are shown as mean ± standard deviation (SD) or median ± interquartile range.
For calcium imaging, the data were post-processed using FEI offline analysis (TILL Photonics GmbH, Germany) and pre-analyzed offline with Image J (Rasband, W.S., Image J, U.S. National Institutes of Health, Bethesda, Maryland, USA). Final analysis and plotting were performed using Origin 2019b software (OriginLab Corporation, Northampton, Massachusetts, USA). To quantify the amplitude of calcium responses, the ratio of the transient fluorescence response to baseline fluorescence was determined (ΔF/F0, the signal-to-baseline ratio; ΔF/F0 = (F-F0)/F0, where F is the calcium-transient peak, and F0 is the averaged baseline fluorescence under resting conditions). Quantitative data are expressed as means ± standard error of mean (SEM), unless otherwise stated. Number of batches and cells within each batch were indicated by n. Significance was assessed with Student’s paired t-test and one-way ANOVA followed by Tukey post hoc test for parametric and non-parametric data, respectively.
P-values ≤ 0.05 were considered significant. Statistically significant differences are shown as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Graphs were created using the GraphPad Prism software (version 8.4.3 for Windows. Total number of statistical units per group is indicated as “n”. Statistical units are depicted as individual data points.
Comments (0)