
Remember me

Burker cell counter (Sigma, cat. n. Z359629)
Centrifuge (Thermo Scientific, model Heraeus Megafuge 1.0)
Coverslip (Sigma, cat. n. Z375357)
Dry ice
Falcon conical tube, polypropylene, centrifuge tubes (50 ml; Falcon, cat. no. 100-0090, 38010)
Falcon conical tubes, polypropylene, centrifuge tubes (15 ml; Falcon, cat. no. 100-0092, 38009)
Filter (0.22 µm Millipore, cat. no. SAMP2GPNK)
Filtered pipette tips (10 μl; BioPoint Scientific, cat. no. 311-4050)
Filtered pipette tips (20 μl; BioPoint Scientific, cat. no. 341-4050)
Filtered pipette tips (200 μl; BioPoint Scientific, cat. no. 351-4050)
Flow Cytometer (Millipore, model Guava® easyCyte 5 HPL Benchtop Flow Cytometer).
Humidified 95% O2/5% CO2 water jacketed incubator, 37 °C (Thermo Scientific, model Forma Series II)
Inverted microscope (Leica, DMIL 090-135.001)
Leica DM2000 fluorescence microscope and DMC5400 camera (Leica)
Line-Gene 9600 system (Bioer Technology)
Low-binding culture dishes (100 mm, Corning, cat. no. 4615)
Microscope slides (75 mm × 25 mm; Corning, cat. no. CLS294875X25)
MidiMACS™ Separator and Starting Kits (Miltenyi biotec, cat. no 130-042-301)
MS Columns (Miltenyi biotec, cat. no.130-042-201)
Multidish (24 well; Falcon, cat. no. 353047)
NanoDrop One Microvolume UV-Vis Spectrophotometers (Thermo Fisher, cat. no ND-ONE-W)
Pipette (1,000 μl; Gilson, cat. no. MSF123602)
Pipette (2 μl; Gilson, cat. no. MSF144801)
Pipette (20 μl; Gilson, cat. no. MSF123600)
Pipette (200 μl; Gilson, cat. no. MSF123601)
Round coverslip (15 mm; Labmaterial, cat. no. 550010-C)
Serological disposable pipette (10 ml; BD Falcon, cat. no. 357551)
Serological disposable pipette (25 ml; BD Falcon, cat. no. 357525)
Serological disposable pipette (5 ml; BD Falcon, cat. no. 357543)
Tissue culture dish (100 mm; Falcon, cat. no. 353003)
Tissue culture dish (150 mm; Falcon, cat. no. 353025)
Tissue culture dish (60 mm; Falcon, cat. no. 353002)
Reagents5X ALL-IN-ONE RT MasterMix (ABM, cat. no. G592)
Accutase 1X (Sigma, cat. no. SCR005)
Adenosine 3′,5′-cyclic Monophosphate, N6,O2′-Dibutyryl-, sodium salt (cAMP, cat. no. 28745-M)
anti-FITC microbeads (Miltenyi biotec, cat. no. 130-048-701)
Antibiotic Solution 100× Liquid, 10,000 U Penicillin and 10 mg Streptomycin per ml in 0.9% normal saline (Himedia, cat. no. A001-100ML)
Antibodies for immunocytochemical analysis (Table 1)
Table 1 Primary and secondary antibodies.Ascorbic Acid 2- Phosphate, 100 mM (Millipore, cat. no 2004011)
Astrocyte Growth Medium (Sigma, cat. no. 821-500)
Astrocyte Growth Supplement (Sigma, cat. no. 821-GS)
B-27™ Plus Supplement (50X) (Gibco, cat. no A3582801)
Brain-derived neurotrophic factor, Human Recombinant Protein (BDNF; PeproTech, cat. no. 450-02)
BrainPhysTM Neuronal Medium (Stem Cell Technologies, cat. 05790)
BrightGreen 2X qPCR MasterMix (ABM, cat. no. G891)
BSA (Gibco, cat. no.16140071)
CHIR-99021- GSK-3 inhibitor (Sigma-Aldrich, cat. no. SML1046-5MG)
DAPI (Thermo Fisher, cat. no. 62248)
DMSO (Sigma, cat no. C6164)
Dulbecco’s modified Eagle medium, high glucose 1× (DMEM; Invitrogen, cat. no. 11965-092)
Dulbecco’s Phosphate buffered saline D-PBS (Sigma, cat. no. D8662)
EDTA (Millipore, cat. no 324503)
Epidermal growth factor (EGF; PeproTech, cat. no. GMP10015)
Fetal Bovine Serum South America Origin, Low Endotoxin level < 0,3 EU/ml (FBS; Microgem, cat. no. RM10432-500ML)
Fibroblast Growth Factor-8 Human Recombinant (FGF-8; Millipore, cat. no. 617103)
FluoroBrite DMEM (Gibco, cat. no. A18967-01)
Formaldehyde solution 37% (Sigma-Aldrich, cat. no. F8775)
Glial-Derived Neurotrophic factor, Human Recombinant Protein (GDNF; PeproTech, cat. no. 45010)
KnockOutTM DMEM/F-12 (Thermo Fisher, cat. no.12660012)
L-Glutamine (Gibco, cat. no. 25030081)
Laminin (Sigma, cat no. L2020)
Matrigel® (Corning, cat. no. 354277)
Methylcellulose MethoCult H4100 (StemCell Technologies, cat. no. 04100)
N-2 Supplement (100×) (Gibco, cat. no 17502048)
Neurobasal™ Medium (Gibco, cat. no. 21103049)
Neurotrophic Factor Human Recombinant Transforming Growth Factor bIII (TGF-b-III; Millipore, cat. no. 617106)
Noggin (Sigma, cat. no. SRP4675)
Non-Essential Amino Acid (NEAA) (Gibco, cat no. 11140050)
Poly (2-hydroxethyl methacrylate) (poly-HEMA; Sigma, cat. no. P3932)
Poly-l-ornithine solution (0.1% (wt/vol) in H2O; Sigma, cat. no. P4957-50ML)
Primer sequences for RT-PCR (Table 2)
Table 2 Primer information for RT-qPCR.Recombinant Human FGF2-basic (bFGF2; PeproTech, cat. no. 100-18B5-0UG)
RNeasy Mini Kit (Qiagen, cat. No. 74104)
SB-431542, TGF-β Receptor Kinase Inhibitor (Med Chem Express, cat. no. HY-10431)
Sonic HedgeHog, Human Recombinant (Shh; Millipore, cat. no. 617102)
Triton X-100 (Sigma-Aldrich, cat. no. X100)
Trypan blue (Sigma Aldrich, St Louis, MO, USA, cat. no. T8154).
Trypsin-EDTA solution (0.25% (wt/vol) trypsin/1 mM EDTA-4Na (1X), liquid; Invitrogen, cat. no. 25200-072)
Water, double distilled (DDW)
Y-27632 10 mM (Sigma, cat. no. 688002)
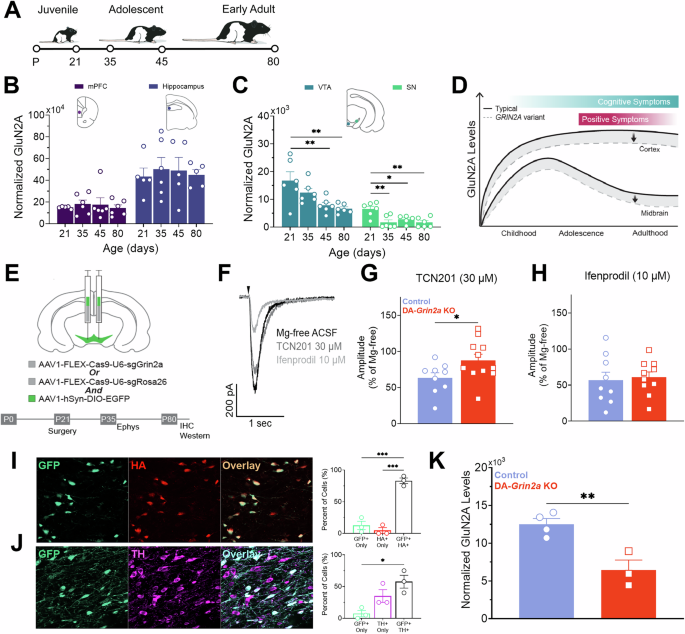
ProcedureIsolation and characterization of Muse cells from skin stromal cellsThe experimental procedures adhered to the guidelines approved by the Ethics Committee of the University of Campania Luigi Vanvitelli (protocol no. 0013954/i). All participants were informed about the nature of the research and provided consent for the use of their biological samples. Skin biopsies were performed using a 1.5 mm punch under medical supervision. The biopsies were processed in vitro to isolate and expand stromal cells. Skin biopsy samples were collected from five donors, aged 25–40 years. Confluent stromal cells and fibroblasts (Fig. 1A) at passage 4 (P4) were harvested using 0.25% trypsin–EDTA and subjected to cell sorting to isolate MUSE cells. The cells were suspended in MACS buffer, which contained 0.5% bovine serum albumin (BSA) and 2 mM EDTA-2H2O in FluoroBrite DMEM. They were then incubated with an anti-human SSEA-3 antibody for 1 h on ice. After this, the cells were washed three times with MACS buffer and centrifuged at 400 g for 5 min. The cells were next incubated with a secondary antibody, anti-rat IgM-FITC, for 1 h on ice and washed three more times. Following that, the cells were incubated with anti-FITC microbeads for 15 min on ice and then washed again with MACS buffer. Magnetic-activated cell sorting (MACS) was used to isolate SSEA-3(+) cells, representing MUSE cells, and SSEA-3(−) non-MUSE cells, according to the manufacturer’s instructions (Miltenyi). The collected SSEA-3 positive cells, which represented MUSE cells, were then cultivated in suspension for 7 days in poly-2-hydroxyethyl methacrylate (pHEMA)-coated petri dishes with a complete medium. The medium consisted of DMEM low glucose, supplemented with 10% FBS, 4 mM L-glutamine, 50 U/ml penicillin–streptomycin, and 2.6% MethoCult [35].
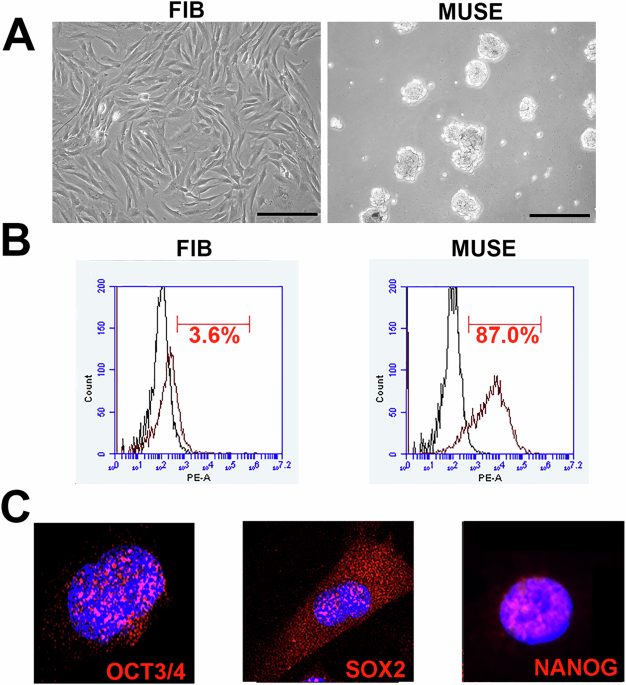
Fig. 1: In vitro characterization of MUSE cells.
A The picture shows a representative image of stromal and fibroblast cells (FIB) and MUSE cells visualized through an inverted microscope (Leica DMIL 090-135.001). 20X magnification. Scale bar 100 µm. B Expression of SSEA-3 surface markers measured by flow cytometry in FIB and MUSE cells. C The pictures are representative images of immunocytochemistry for stemness markers: OCT3/4, SOX2 and NANOG, performed on MUSE cells. The nuclei were counterstained with DAPI (blue). 20X magnification.
MUSE cells were cultured on pHEMA for 7 days to select and enrich the population of MUSE stem cells (Fig. 1A). When grown in suspension, MUSE cells overexpress stemness markers such as OCT3/4, SOX2, and NANOG. At the end of the 7 days, the MUSE cells were characterized for the expression of the surface marker SSEA-3 using flow cytometry and for the expression of stemness markers using immunocytochemistry (Fig. 1B, C).
MUSE cells were washed with PBS and incubated with anti-SSEA-3 PE-conjugated antibody, following the manufacturer’s instructions. After 30 min of incubation with the antibodies in the dark at room temperature, the cells were washed again with PBS and resuspended in MACS buffer for analysis using a Guava easyCyte flow cytometer. Data analysis was performed following standard procedures using easyCyte software, with at least 5000 cells per sample analyzed, and gating was done based on forward scatter (FSC) and side scatter (SSC) channel signals (Fig. 1B).
cells were seeded on coverslips in a 24-well plate overnight and then fixed with a 4% formaldehyde solution for 15 min at room temperature. After fixation, the cells were washed three times with PBS and permeabilized with 0.3% Triton X-100 in PBS for 15 min. After another PBS wash, the cells were incubated for 1 h at room temperature in a blocking solution consisting of 0.1% Triton X-100 and 5% bovine serum in PBS. The cells were then incubated with primary antibodies (OCT3/4, SOX2, NANOG; see Table 1) in blocking solution overnight at 4 °C, following the manufacturer’s instructions for each antibody. The next day, after three washes with PBS (5 min each), the cells were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature in the dark. After three more PBS washes (5 min each), the coverslips were mounted. Nuclear staining was performed using a DAPI mounting medium, and micrographs were taken using a fluorescence microscope (Leica)(Fig. 1C).
Neural progenitor differentiation Preparation of Matrigel-coated platesMatrigel is used to pre-coat cell culture dishes and provide an attachment surface for feeder-free human cells to replicate on. To avoid warming up Matrigel stock and solidifies at temperatures above 10 °C, special care is required during its preparation.
1.Defrost Matrigel: 1 day before use, defrost the matrigel at 4 °C overnight on ice or in a cold chamber to maintain the temperature during the whole process.
2.Prepare the coating solution: Dilute Matrigel 1× at a ratio of (1:50) using pre-cooled DMEM/F12 in a Falcon tube and transfer the desired volume to the plate. Add 5 mL in 100 mm plates and 2 mL in 60 mm plates. Gently shake the mix with a pipette, avoiding the formation of bubbles. Next, distribute it evenly on each culture plate. If necessary, gently tap the plate on the work surface to make sure the liquid covers the entire surface evenly. Incubate the Matrigel-coated plates at 37 °C and 5% CO₂ for at least 1 h.
3.Remove the solution: After incubation, aspirate excess liquid Matrigel from the plates and immediately add cells or culture medium to the plate to prevent the Matrigel coating from drying out.
4.Preparation before use: Just before use, aspirate the solution from the coated cultureware and add the Neuronal Differentiation Medium to the cultureware.
Induction of Neural differentiation (day 1 to 13)MUSE cells grown in suspension on pHEMA for 7 days are collected using a pipette into a 15 ml Falcon tube. The plate is washed thoroughly with PBS to collect all the cells, and the liquid is added to the 15 ml Falcon tube. Subsequently, the cells are centrifuged for 5 min at 300 g at room temperature. The supernatant is discarded, and the pellet is resuspended in 1 mL of Neural Induction Medium 1 (NIM1: DMEM/F12, 50 U/ml penicillin–streptomycin, N2 1%, B27 4%, L-Glut 1 mM, BSA 1 mg/mL, NEAA 1%, SB431542 TGF-β receptor inhibitor 10 µM, Noggin 200 ng/mL, Laminin 1 µg/mL, Table 3). The cells are seed on Matrigel coated dish, subsequently other 7 ml of NIM1 are added to the dish. The cells are then collected and plated at a density of 1 × 105 on a 100 mm Corning plate coated with Matrigel, which has been prepared in advance. After plating the cells on Matrigel, an additional 8 mL of fresh NIM1 is added. Half of the medium, approximately 4 mL, is removed and replaced with freshly prepared NIM1 every 3 days until day 7.
Table 3 Media composition for a final volume of 10 ml.During the first week, the cells exhibit proliferative capacity and can reach 80-90% confluence. Upon reaching day 7, the NIM1 medium is completely removed and replaced with Neural Induction Medium 2 (NIM2: DMEM/F12, 50 U/ml penicillin–streptomycin, N2 1%, B27 4%, L-Glut 1 mM, BSA 1 mg/mL, NEAA 1%, Laminin 1 µg/mL, Table 3) for 5 days. The medium is completely changed every day to ensure the removal of any inhibitors present in NIM1, until day 13 is reached. At this point, the cells should have reached 80–90% confluence (Fig. 2A).
Fig. 2: Morphological progression of MUSE cells toward neural progenitors.
A Neural progenitor cells on day 13. B Neurosphere on day 16 on low-binding plates C Neurosphere adhering to Matrigel-coated plate. D Neuroprogenitor cells on day 30. Cells are visualized through an inverted microscope (Leica DMIL 090-135.001). 20X Magnification. Scale bar 100 µm.
Neurosphere formation and neural progenitor propagation (day 13 to 30)On day 13, the cells have reached the first stage of neural differentiation, adopting a more elongated and branched morphology. The NIM2 medium is completely removed, and the plates are washed 3 times with PBS to thoroughly remove the medium. The cells are treated with Accutase 1× (3 ml for 100 mm plate) for 5 min at 37 °C in 5% CO₂. After the treatment, the cells detach from the plastic and are suspended in the petri dish. The cells are collected with 2 ml of FBS using a pipette to ensure that all the cells are gathered and then placed into a 15 ml Falcon tube. Subsequently, the cells are centrifuged for 5 min at 300 g at room temperature, the supernatant is removed, and the cells are resuspended in 1 ml of Neural Progenitor Medium (NPM: DMEM/F12, 50 U/ml penicillin–streptomycin, N2 1%, B27 4%, L-Glut 1 mM, Laminin 1 µg/mL, EGF 20 ng/mL, FGF2 20 ng/mL, Y-27632 ROCK inhibitor 1 µM, Table 3). The cells are plated on low-binding 100 mm plates at a density of 1 × 10⁶ for 3 days. After plating the cells on low binding plates, an additional 8 mL of fresh NPM is added.
After 3 days of growth in suspension, the cells form neurospheres (Fig. 2B). The cells create small clusters composed of multiple cells, which can later be collected and differentiated. At the end of the three days in suspension, the cells are collected with a pipette and placed into a 15 ml Falcon tube. The cells are centrifuged for 5 min at 300 g at room temperature, the supernatant is discarded, and the pellet is resuspended in 1 ml of Neural Progenitor Medium (NPM). The cells are counted and plated at a density of 5 × 105 cells on a plate coated with Matrigel, which has been prepared in advance following the previous protocol. An additional 7 ml of NPM is added, and the cells are incubated at 37 °C with 5% CO₂ in a humidified atmosphere (Fig. 2C).
For the neurospheres plated on Matrigel with neural progenitor medium (NPM), half of the medium (approximately 4 mL) will be replaced twice a week with fresh medium until the cells reach confluence. In about 2–4 weeks, the cells will reach confluence and the neural progenitor stage (Fig. 2D). The cells will exhibit the following characteristics: an elongated or fusiform shape with thin processes extending from the cell body, which may become more numerous and branched as the cells differentiate into neurons or glia; a compact cell body and dynamic morphology, acquiring a more complex and branched structure as differentiation progresses. At this point, the neural progenitors can be harvested and used in experiments to characterize expression markers and analyze biological features. The progenitors can also be harvested and cryopreserved in DMSO and serum for future characterization or experimentation. Finally, the progenitors can be differentiated into GABAergic and glutamatergic neurons or into astrocytes (see protocols below).
Neurosphere plating issuesAfter the formation of neurospheres, it is common to lose part of the cell population when replating, as some cells may not adhere to the Matrigel-coated plate after the suspension step. This can occur because the neurosphere clusters do not adhere well to the substrate and are unable to readapt immediately. To address this issue, it is important to pipette the neurosphere cell pellet carefully and gently after centrifugation to break up the clusters and obtain single cells in suspension. Use a P200 pipette and gently rest the pipette tip on the side of the Falcon tube, avoiding rapid pipetting that could damage the cells. Then, replate the cells evenly onto the new Matrigel-coated plate. It is also good practice to start with a sufficiently high number of cells to minimize excessive cell loss during this phase.
After collecting the cells from suspension growth and plating them on Matrigel, the cells must reach confluence before starting the final step of neural differentiation. For this reason, the cells should no longer be trypsinized or replated. At this stage of the protocol, it is advisable to carefully plan the experiments and analyses to be conducted at the end of the differentiation protocol. Therefore, the cells should be plated in sufficient numbers and on appropriate plates to carry out the subsequent experiments.
Use of inhibitor in Neural differentionIn neural differentiation in vitro, the use of SMAD pathway inhibitors such as Noggin, Y-27632 and SB431542 is essential to direct stem cells toward neural differentiation. The SMAD pathway is mostly activated by signals belonging to the families of transforming growth factors (TGF-β) and bone morphogenetic proteins (BMPs), which play a crucial role in the control of cell fate, including processes such as proliferation, differentiation, and apoptosis [36].
In particular, Y-27632 is an inhibitor of Rho-associated protein kinase (ROCK), a kinase involved in regulating the cytoskeleton by modulating actin polymerization. Rock is used to enhance cell survival, facilitate neurite outgrowth, and stabilize cell adhesion during the transition to a neural lineage reducing the risk of apoptosis induced by cell detachment (known as anoikis), which often occurs during stem cell culture and differentiation [37].
GABAergic and Glutamatergic differentiation (day 30 to 50)The medium is completely removed from the neural progenitors, and the cells are washed 3 times with PBS. The medium is then replaced with Neuronal Differentiation Medium (NDM: BrainPhys Medium, 50 U/ml penicillin–streptomycin, N2 1%, B27 4%, Laminin 1 µg/mL, BDNF 20 ng/mL, GDNF 20 ng/mL, Table 3).
The cells were cultured in NDM for 20 days to allow differentiation into GABAergic and Glutamatergic neuronal phenotypes. Every 3 days, half of the NDM was removed and replaced with freshly prepared medium to avoid degradation of the growth factors, which have a short half-life.
At the end of the 20 days, a portion of the mature neurons exhibited GABAergic or Glutamatergic phenotypes (Fig. 3A). GABAergic neurons are generally smaller and less branched compared to Glutamatergic neurons. They have a higher number of short, branched dendrites, often exhibiting complex dendritic branching. Glutamatergic neurons tend to have a more complex and branched morphology, with extended dendritic trees and a higher density of dendritic spines, corresponding to their role as the main excitatory neurons in the brain.
Fig. 3: Differentiation of MUSE cells into neuronal and glial subtypes.
A GABAergic and Glutamatergic differentiated cells. B Committed Astrocytes. C Dopaminergic differentiated cells. Cells are visualized through an inverted microscope (Leica DMIL 090-135.001). 20× Magnification. Scale bar 100 µm.
At the end of the differentiation process, the neurons can be collected for analysis of protein expression through immunocytochemistry or western blot, RNA expression analysis via RT-qPCR, or electrophysiological analysis of the neurons.
Neural committed cells were detached from the Matrigel-coated plates using Accutase 1× (3 ml for 100 mm plate) for 5–7 min at 37 °C with 5% CO₂. Once the cells were detached from the plastic, they were suspended in the petri dish. The cells were then collected with 2 ml of FBS using a pipette to ensure complete collection and transferred into a 15 ml Falcon tube. The cells were centrifuged at 300 g for 5 min at room temperature, and the supernatant was discarded. The cell pellet was resuspended in PBS to wash and remove any residual culture medium. Following another round of centrifugation, the pellet was resuspended in lysis buffer for RNA extraction. Alternatively, the cells can be resuspended in NDM and plated on poly-ornithine and laminin-coated plates for immunocytochemistry experiments.
Astrocyte differentiation (day 30 to 60)The neural progenitors generated following the previously outlined protocol can also be used for astrocyte induction. After three washes with PBS to thoroughly remove all residual medium, 8 ml of Astrocyte Differentiation Medium (ADM: Astrocyte Growth Medium, Astrocyte Growth Supplement, 50 U/ml penicillin–streptomycin, FBS 2% fetal bovin serum, Table 3) are added to the neural progenitors for 30 days. During this period, half of the medium is replaced with fresh medium every three days. To avoid passaging or trypsinizing the cells, seed them at a low density. This comprehensive protocol enables the differentiation of MUSE cells into fully mature astrocytes. By the 30th day, a portion of the cells have developed a characteristic astrocytic phenotype (Fig. 3B). In vitro, these astrocytes typically display a star-shaped morphology with multiple branched processes extending from the cell body. These processes aid in synaptic support and the maintenance of homeostasis in neuronal environments. The cells exhibited also markers typical of mature astrocytes reflecting their functional and morphological maturity after the differentiation protocol.
Upon completion of the differentiation process, the astrocytes can be harvested for protein expression analysis using immunocytochemistry or western blot, or for RNA expression analysis through RT-qPCR (as detailed below).
Pre-dopaminergic neurons differentiation (day 1 to 14)MUSE cells cultured in suspension on pHEMA for 7 days are collected with a pipette and transferred into a 15 mL Falcon tube. The plate is thoroughly washed with PBS to ensure all cells are collected, and the washing liquid is added to the same tube. The cells are then centrifuged at 300 g for 5 min at room temperature. After discarding the supernatant, the cell pellet is resuspended in 1 mL of Dopaminergic Induction Medium 1 (DIM1: DMEM-F12, pen-strep 50 U/mL, N2 1%, L-glutamine 2 mM, SB431542 (TGF-β inhibitor) 10 µM, Noggin 200 ng/mL, Table 3). The cells are plated onto a 100 mm dish coated with Matrigel (see protocol above), and an additional 7 mL of DIM1 is added. The cells are cultured in this medium for 5 days, with the medium being replaced with fresh DIM1 every two days. On the fifth day, the medium is removed, and the cells are washed twice with PBS to completely eliminate residual medium and any factors. Then, 8 mL of Dopaminergic Induction Medium 2 (DIM2: DMEM-F12, pen-strep 50 U/mL, N2 1%, B27 4%, L-glutamine 2 mM, SB431542 10 µM, Noggin 100 ng/mL, SHH 100 ng/mL, FGF8 100 ng/mL, CHIR99021 (GSK-3 inhibitor) 3 µM, Table 3) is added. The medium is replaced every two days with fresh DIM2.
On the tenth day, the medium is fully removed, and the cells are washed with PBS. Dopaminergic Induction Medium 3 (DIM3: Neurobasal Medium, pen-strep 50 U/mL, N2 1%, B27 4%, Laminin 1 µg/mL, Noggin 100 ng/mL, SHH 100 ng/mL, FGF8 100 ng/mL, CHIR99021 3 µM, Table 3) is added. The cells are cultured in this medium until day 14, with the medium being replaced every two days. During this phase, the cells will continue to proliferate, reaching the neural precursor stage with a predisposition toward differentiation into dopaminergic neurons.
Dopaminergic neurons differentiation (day 14 to 30) Preparation of poly-L-ornithine and laminin-coated platesNeural progenitor cells should be cultured on poly-L-ornithine and laminin-coated culture ware before starting terminal differentiation. The following steps are recommended:
1.Prepare the coating solution: Dilute the Poly-L-Ornithine solution 0.1 mg/mL (1:10) with PBS to achieve a final concentration of 10 µg/mL. Add enough volume of the Poly-L-Ornithine solution to fully cover the surface of the cultureware (5 mL in 100 plate). Incubate at room temperature for 2 h.
2.Prepare the Laminin: Thaw the laminin, provided at 1 mg/mL, on ice. Using sterile 1X PBS, dilute the laminin to a final concentration of 10 µg/mL.
3.Remove the Poly-L-Ornithine solution: Aspirate the Poly-L-Ornithine solution. Add sufficient volume of the 10 µg/mL laminin solution to cover the entire surface of the cultureware (5 mL in 100 plate). Incubate at room temperature for 2 h.
4.Storage of coated plates: Coated plates and flasks can be stored in the laminin solution at −20 °C for up to six months. Wrap the plates in plastic wrap before storing them at −20 °C.
5.Preparation before use: Just before use, aspirate the laminin solution from the coated cultureware and wash once with 1× PBS. Add the Neuronal Differentiation Medium to the cultureware. Do not allow the plates or wells to dry out, as this may result in uneven cell attachment.
On day 14, the cells are washed twice with PBS, and the medium is replaced with Dopaminergic Differentiation Medium (DDM: BrainPhys medium, pen-strep 50 U/mL, N2 1%, Laminin 1 µg/mL, BDNF 20 ng/mL, GDNF 20 ng/mL, TGF-βIII 1 ng/mL, ascorbic acid 0.2 mM, cAMP 0.5 mM, Table 3). The cells are cultured in this medium until reaching confluency (around day 20), with the medium being refreshed every 3 days. At confluency, on day 20, the cells are detached from the Matrigel-coated plates using Accutase (3 mL) for 5–7 min at 37 °C with 5% CO₂. Once detached, the cells are suspended in the Petri dish, collected in 1 mL of DDM, and counted. The cells are centrifuged at 300 g for 5 min and resuspended in the appropriate volume of DDM. Subsequently, the cells are seeded onto Poly-L-ornithine/Laminin-coated plates (see protocol above) at a desired concentration, depending on the planned experiments and analyses.
The neural cells are grown for approximately 10 days in DDM to allow for the final differentiation into dopaminergic neurons (Fig. 3C). The medium is replaced with fresh DDM every 3 days. At the end of differentiation, a portion of the cells will exhibit the phenotypic characteristics of mature neurons, such as: Morphological features: Dopaminergic neurons typically display a well-defined morphology, characterized by the presence of long, branched dendrites and a single axon. The dendritic arborization allows for extensive synaptic connections, facilitating communication with other neurons. Neurite outgrowth: Following differentiation, these neurons often extend long neurites that can branch extensively, which is crucial for their functional connectivity. Cell body shape: The cell bodies of dopaminergic neurons tend to be relatively larger and are often round or oval in shape, containing prominent nuclei.
Upon completion of the differentiation process, the dopaminergic neuron can be harvested or fixed for protein expression analysis using immunocytochemistry or western blot, or for RNA expression analysis through RT-qPCR (as detailed below).
Use of inhibitor and growth factor in dopomergic neuron differentionIn dopaminergic differentiation, the use of specific inhibitors is essential to guide stem cells toward a neuronal fate and promote the formation of dopaminergic neurons. In particular, SB431542 is a specific inhibitor of TGF-β (transforming growth factor beta) receptor kinases, specifically ALK4, ALK5 and ALK7 receptors. Inhibition of Activin/Nodal/TGF-β signaling pathways, which normally induce maintenance of the undifferentiated state or promote other differentiation pathways such as mesodermal or endodermal, in this case promotes neural fate [36].
CHIR99021 is a selective inhibitor of glycogen synthase kinase-3 (GSK-3), in this way preventing phosphorylation of β-catenin, which would normally be degraded. Inhibiting β-catenin degradation allows its accumulation in the cytoplasm and subsequent transport into the nucleus, where it can activate target genes that promote neuronal differentiation [38].
Noggin is a natural protein that works as an extracellular antagonist of BMP. It binds to BMP molecules before they can interact with their receptors, thereby preventing activation of the BMP pathway, thus promoting differentiation toward neuronal cells [39].
Together, these inhibitors create an environment conducive to the production of dopaminergic neurons by blocking pathways that would otherwise divert differentiation toward other cell types.
Immunocytochemistry (ICC) analysis of differentiated cellsTo accurately assess the different stages of differentiation using immunocytochemical analysis, it is essential to plate cells on Poly-L-ornithine and Laminin-coated plates (see protocol above). When Matrigel is used as a coating, it can interfere with antibody or fluorescent probe penetration, especially if applied too thickly. Additionally, Matrigel can exhibit autofluorescence, particularly when exposed to light for extended periods. For these reasons, the use of Poly-L-ornithine and Laminin as coating materials is often preferable to Matrigel. Poly-L-ornithine is a cationic polymer that enhances cell adhesion, particularly for neural cell types. Its positive charge interacts with the negatively charged molecules on the cell surface, strengthening the attachment. Laminin, on the other hand, is an extracellular matrix protein that specifically promotes the adhesion and survival of various cell types, including neurons and stem cells. It also supports cell growth and differentiation. By using Poly-L-ornithine and Laminin, the complexity of Matrigel, which contains growth factors and proteins that may interfere with antibody staining, is avoided. Together, Poly-L-ornithine and Laminin provide a more controlled, reproducible substrate suitable for a wide range of cell types. This is particularly advantageous in experiments where maintaining cell morphology and minimizing biological variability are critical, making them ideal for many immunocytochemical assays.
Cells were seeded on coverslips in a 24-well plate for 2 days in the specific related media (NDM, DDM, ADM) and then fixed with 4% formaldehyde for 15 min at room temperature. After fixation, the cells were washed three times with PBS and permeabilized with 0.3% Triton X-100 in PBS for 15 min. Following another PBS wash, the cells were incubated at room temperature for 1 h in a blocking solution containing 0.1% Triton X-100 and 5% bovine serum in PBS. Primary antibodies in blocking solution (as listed in Table 1) were added, and the cells were incubated overnight at 4 °C. All antibodies were used according to the manufacturer’s guidelines. The next day, the cells were washed three times with PBS (5 min each), and then incubated for 1 h at room temperature in the dark with secondary antibodies conjugated to fluorophores. Afterward, the cells underwent three additional 5 min washes with PBS, and the coverslips were mounted. Nuclear staining was performed using DAPI mounting medium, and images were captured using a fluorescence microscope (Leica). The percentage of positive cells was determined by counting at least 500 cells across different microscope fields (Fig. 4).
Fig. 4: Immunocytochemistry analysis of lineage-specific markers after neural differentiation.
A Representative picture of ICC staining after neural differentiation. NESTIN was used as Neural Progenitor Markers; MAP2 and TUBB3 were used as a neuron marker; GABA was used as a Gabaergic neuron marker; TH was used as Dopaminergic neuron marker; GLS and VGluT1 were used as Glutamatergic marker; GFAP and S100-B were used as astrocyte markers. The nuclei were counterstained with DAPI (blue). 20X magnification. Scale bar 20 µm. B Differentiation efficiency of MUSE cells into various neural cell types. The bar graph illustrates the percentage of MUSE cells that successfully differentiated into committed neurons, GABAergic neurons, glutamatergic neurons, dopaminergic neurons, and astrocytes. Data are presented as mean ± SEM (n = 3).
RT-qPCR analysis of differentiated cellsTotal RNA was isolated from cell cultures using the RNeasy Mini Kit (Qiagen), following the protocol provided by the manufacturer. The RNA’s quality and concentration were evaluated using a Nanodrop spectrophotometer. Primer pairs for real-time RT-qPCR reactions were designed based on mRNA sequences retrieved from the Nucleotide Data Bank (National Center for Biotechnology Information) using Primer Express v. 3.0 software (Applied Biosystems). The primer sequences can be found in Table 2. cDNA synthesis was carried out using the 5X ALL-IN-ONE RT MasterMix. GAPDH cDNA regions were used as controls, and the real-time PCR was performed on a Line-Gene 9600 system (Bioer Technology). The reactions followed the manufacturer’s protocol, utilizing BrightGreen 2X qPCR MasterMix. For data analysis, the 2−ΔΔCT method was applied for relative quantification in the real-time PCR experiments (Fig. 5).
Fig. 5: mRNA expression levels of neural differentiation markers.
The histograms show the quantitative RT-PCR analysis of neural progenitors’ markers, GABAergic, glutamatergic and dopaminergic markers, astrocytes markers. The mRNA levels were normalized to GAPDH mRNA expression, which was selected as an internal control. Data are expressed as fold changes with standard error (n = 3). For each gene, the expression level of not differentiated MUSE (ND-MUSE) is set as the baseline (the arbitrary value is 1).
Comments (0)