With respect to East Austria, which has approximately four million inhabitants, our IASU is the only dedicated institution that has reviewed a total of approximately 150 paediatric patients over the past decade.
Owing to a continuous transition to adult care, numbers are always fluctuating, explaining our actual count of 112 patients (125 including patients with VUS) at the last cross-sectional reference date. However, despite these fluctuations, there has been an overall constant increase in patient numbers over the past 10 years, which may be in part due to a 15% increase in the population. Moreover, this is founded on the burden of responsibility for patients with potentially life-threatening diseases and—at least in the beginning—the lack of genetic analysis capacity outside a tertiary care centre, both leading to a trustful cooperation with other hospitals and practitioners in East Austria and thereby ensuring data consistency over the past decade. Notably, our survey, performed among paediatric cardiologists, confirmed consistent referral adherence.
Despite these efforts, the difference between our “Real World” prevalence and the expected numbers according to the published prevalence data is still significant. However, our prevalence depends on a suspicion of diagnosis due to clinical reasons or family history, which is a completely different approach from the available data obtained from screening studies. Furthermore, molecular autopsies are not performed routinely after sudden death, particularly in children. Addressing this issue in our country would not only influence prevalence data but also improve preventive personalized measures to reduce the risk of malignant arrhythmias and sudden death among the relatives of victims.
LQTS
For LQTS, the paediatric prevalence is based on the findings of a community-based screening program performed by Schwartz et al. 15 years ago [19]. In this pivotal study, the authors revealed a prevalence close to 1:2000 for phenotypically and/or genetically proven LQTS, which remains the highest published prevalence in the Caucasian population thus far. The same group analysed the cost-effectiveness of nationwide screening programmes and reported that neonatal ECG screening was feasible and cost-effective for preventing unnecessary deaths during infancy and childhood [20]. Furthermore, the probability of a diagnosis of LQTS in a school-based screening program performed in Japan was found to be 1:3300 for subjects aged 6 years and 1:1000 for those aged 12 years [21]. However, according to a study published in 2010, most paediatric cardiologists are still sceptical of ECG screening for LQTS [22], and screening programmes are not established in most countries. Our findings obtained under “Real World” conditions describe the opposite approach to diagnosing these conditions. However, our prevalence of 1:13,000 is almost sevenfold lower than that published by Schwartz et al. although we used the same inclusion criteria (genetic testing if repetitive ECGs with QTc between 460 and 480 ms or higher) and can ensure extensive referral adherence. One of the possible reasons might be the age-dependent detection rate caused by the lower frequencies of ECGs conducted in infancy and early childhood, with the consequence that some affected infants and younger children escape early diagnosis by “Real World” approach.
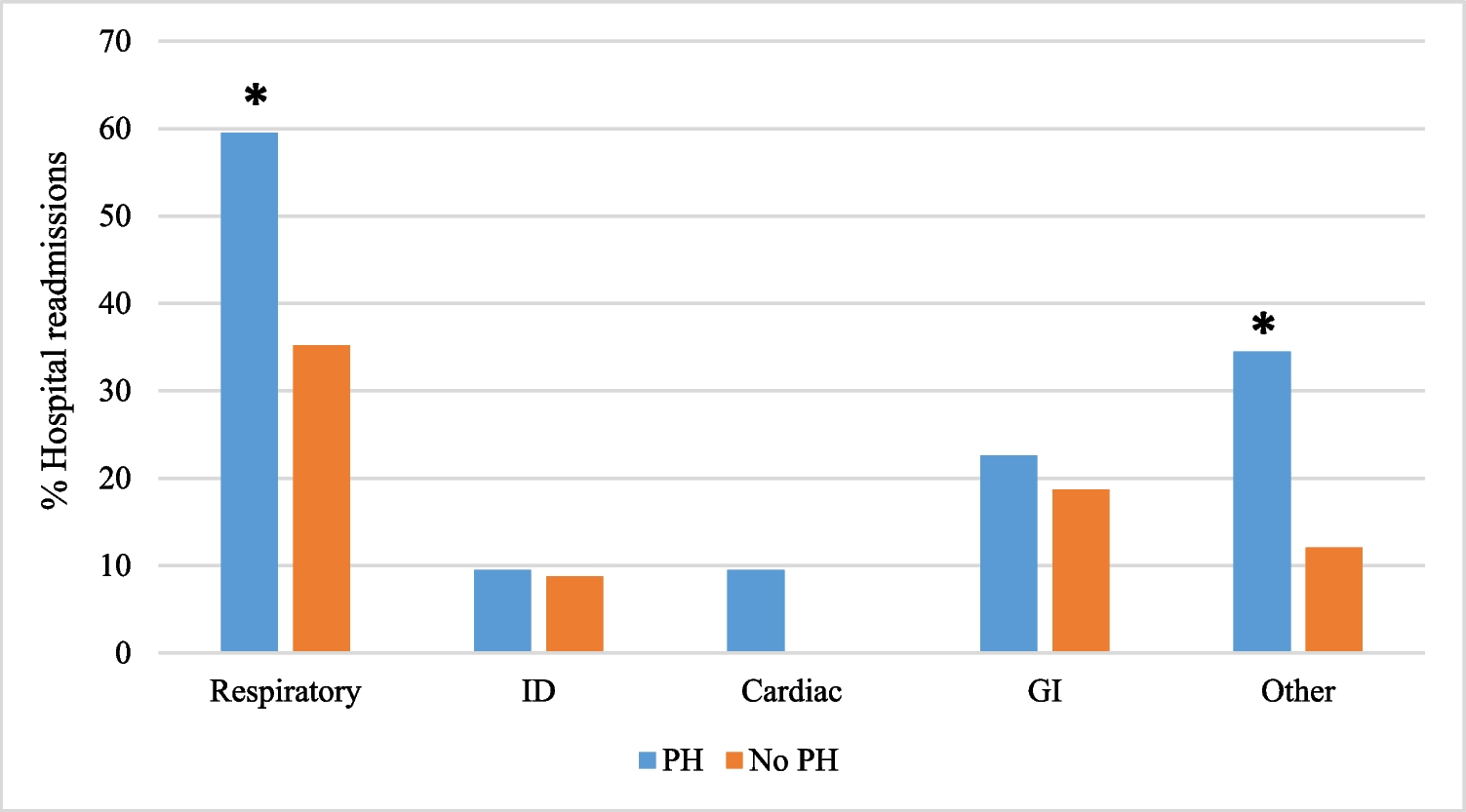
A recently published study of cardiovascular preparticipation screening (PPS) in 22,000 consecutive young competitive athletes revealed that compared with a single-only PPS, a repeat cardiovascular evaluation during an 11-year study period increased the diagnostic yield of cardiovascular disease at risk of SCD and that the peak diagnostic yield is among children older than 12 years [23]. This finding is in accordance with our findings, revealing a higher frequency of LQTS detection in older children, most likely due to an increasing number of recorded ECGs with increasing age (Fig. 2).
Finally, given the variation in the prevalence of LQTS resulting from different diagnostic approaches for a disease with a possibly high burden even in childhood, it seems reasonable to rethink the value of LQTS screening or staged screening programmes in adolescence to detect asymptomatic probands.
Genetic testing at our institution was found to have a relatively high diagnostic yield of 94%, which might be explained by the strict selection of patients. Ideal screening should produce as few false-positive results as possible while detecting probands who are at risk. Given that the threshold QTc interval used for diagnosis is the primary determinant of test sensitivity and specificity, a false-positive rate for those with a QTc interval of > 460 ms is less than 1 in 1000 but 4 in 1000 for those with a QTc interval of > 450 ms [4]. At our institution, genetic testing is initiated in the case of repeated QTc intervals > 460 ms, which certainly contributes to the high diagnostic yield. Notably, we regularly measure the QT interval manually and calculate the QTc with Bazett’s equation to take sinus arrhythmia into account and thereby avoid false-positive results based on automatically derived QTc intervals, which may contrast with other institutions with lower diagnostic yields of genetic testing (30% to 70%) [24, 25].
Furthermore, genetic testing revealed eight carriers of VUS, which is a challenge in clinical practice, most likely raising more questions than answers. Interestingly, most of them presented significant phenotypic expression, with QTC intervals ≥ 500 ms (Supplementary Table 1). Despite the unclear significance of the mutations, we consider these patients at risk and follow them like genetically positive patients, given that some variants might be reclassified in the future to be pathogenic.
BrS
BrS is described as a Mendelian syndrome with an autosomal dominant inheritance pattern and incomplete penetrance [26]. Different studies propose varying adult prevalences ranging from 1:5000 to 1:2000 [27, 28], depending on region and ethnicity. Its prevalence is higher in Asia and the Middle East, where estimates range between 1:270 and 1:625 [29]. The current literature on paediatric prevalence is restricted to Japanese school-based screening programmes, suggesting much lower numbers in children despite growing evidence of disease onset early in childhood [30]. As BrS is clinically and phenotypically expressed mainly in the third or fourth decade of life, most children are identified by family cascade screening [11]. As such, patients enrolled in our study were diagnosed either in the context of extended family screening or, less commonly, after presenting with a Brugada-like ECG. Our prevalence of 1:22,000 obtained under “Real World” conditions is four to ten times lower than that in adults and may result from incomplete referrals of family members after diagnosis of an adult index patient. In recent years, we have offered genetic analyses to all family members. While a SCN5A mutation can be found in only 20–25% of the adult population with BrS [31], the prevalence of a gene mutation in paediatric patients is higher, reaching 58.1% [32], which is in accordance with our current positive yield of 66%.
ARVC
ARVC should be considered in adolescents or young adults who present with symptoms of palpitations, PVCs, syncope, or aborted SCD. The adult prevalence of ARVC is estimated to be between 1:5000 and 1:2000 [33, 34], whereas our paediatric prevalence was 1:43,000, with a median patient age of 13 years (1–18 years). While age-related penetrance is evident in ARVC, with the highest incidence occurring between the ages of 30 and 40 years, it is noteworthy that no comprehensive studies have been conducted to systematically assess ARVC epidemiology during childhood [35].
Although ARVC phenotypically presents as “cardiomyopathy”, resulting in high alertness among adult cardiologists, the diagnosis in our patients was made mainly by a paediatric cardiologist, including adult siblings of children who were referred to us after unexplained SCD of a family member. Our significantly lower prevalence of BrS and ARVC than in adults is attributed to the fact that in common practice, only patients with clinical features or family history can be diagnosed. As shown in our analysis, particularly for BrS and ARVC, family history is the key to diagnosis in children. This highlights the necessity of intensive collaboration with adult cardiologists, including even more generous referrals of paediatric family members, not only in the case of an IAD diagnosis but also after unexplained SCD of an adult family member. In addition, as disease penetrance increases with age and peaks in young adulthood, our numbers might be equally correct. However, given the paucity of paediatric prevalence studies, we sought to compare our results to available adult data to highlight that higher detection rates should be expected, predominantly in adolescence or young adulthood.
CPVT
The literature-reported estimates of overall CPVT prevalence are 1:10,000 or less [6, 36, 37]. The population-based paediatric prevalence might be uncertain, as CPVT is difficult to diagnose and easily missed, especially in patients in whom SCD is the first manifestation at a young age.
Our prevalence of 1:368 000 is very low, and the gap in published numbers might illustrate the difficulties of suspecting this diagnosis, as ECG at rest will generally be normal. Presumably, we still need to increase alertness for this disease among primary health care providers and paediatric cardiologists in cases of unclear fainting or syncope associated with stressful situations or activities, as CPVT is associated with an annual near-fatal and fatal event rate of 1.9%, decreasing to 0.8% in treated patients [38].
Limitations of this analysis include the small number of patients and that the study was conducted only in the eastern part of Austria. The design of our study implies that our prevalence is dependent on the alertness and cooperation of referring colleagues in the paediatric and adult fields. Although we can largely confirm referral adherence in our catchment area, we cannot exclude the loss of some patients. However, even if a substantial number of additional patients were treated by other colleagues or hospitals not reached by our survey, our prevalence numbers would still be very low. Comparisons of our “Real World” prevalence with available data should be performed with caution because of the different study designs used.
As a tertiary care centre, we are dependent on referrals. Practitioners and referring hospital colleagues were not referring probands with QTc values below 460 ms without other symptoms, thus, there might be some LQTS patients who did not reach our IAD tertiary unit. Furthermore, probands with a Schwartz score between 1.5 and 3 are very likely to remain undiagnosed, as repolarization abnormalities or bradycardia in an ECG conducted in a general paediatric setting are rarely the reason for referral, particularly in the absence of symptoms or family history. We excluded probands with VUS from the statistical analysis, which additionally decreased our prevalence data.

Comments (0)