
Remember me
Peripheral nerve sheath tumours (PNST) in schwannomatosis (SWN) comprise schwannoma (Sw) and hybrid neurofibroma/schwannoma (HNS). The second tumour type, the hybrid variant, is recognized on the basis of relatively recent insights into the neuropathological characterization of SWN-associated neoplasms. However, it is ONLY as recent as the updated diagnostic criteria proposing the term “schwannomatosis” as an umbrella term for neurofibromatosis type 2 (NF2) and SWN, that novel classification of SWN based on disease-causing genes was proposed [1]. It is important to know this history to correctly interpret publications before 2022 that differentiated between NF2 and SWN.
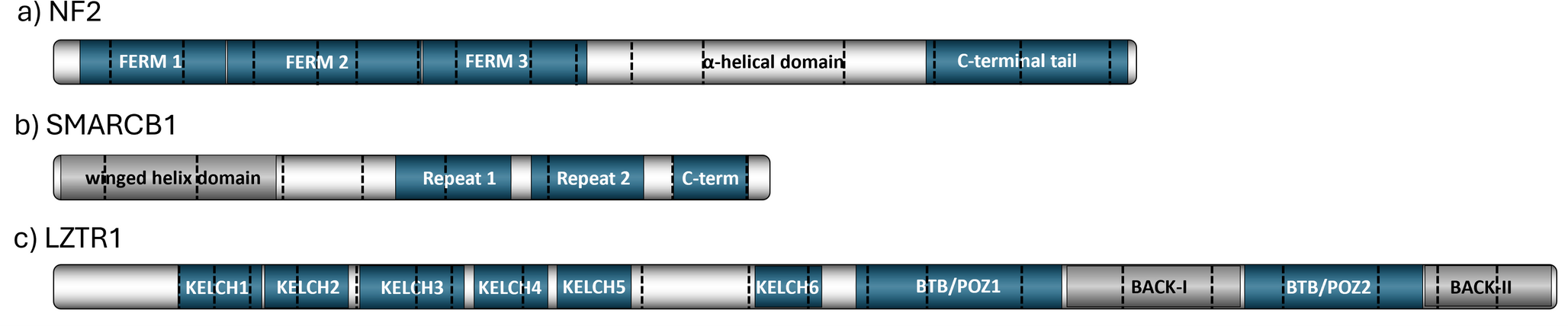
The 2022 international consensus recommendation correctly pointed out that misdiagnosis of Sw and HNS as neurofibroma (Nf) has led to numerous incorrect diagnoses of neurofibromatosis type 1 (NF1). Accurate diagnosis is particularly important in forms of SWN not related to NF2 variants, such as LZTR1- or SMARCB1-associated SWN, or SWN-NEC (not elsewhere classified), as these subtypes are defined by fewer major diagnostic criteria compared with NF2-related SWN. Consequently, the histopathological identification of Sw or HNS plays a significantly more critical role in the diagnostic process and has thus been explicitly included in the official recommendation tables.
HNS have been reported in a few studies so far. It took 80 years (Sw was first recognized histologically in 1910 by Jose Verocay) for HNS to be distinguished from conventional Sw by Feany and co-workers, who described a series of 9 cases in 1998 [2]. In 2012, a clear association of HNS to neurofibromatosis type 1 and especially to SWN (certainly back then NF2-SWN and SWN-NEC) was described by Harder and co-workers, who also first elucidated the molecular genetics in a subsequent study [3, 4]. Thereafter HNS was integrated into tumour WHO classifications [5, 6]. In 2016 another detailed histological study of HNS in 11 cases showed clear evidence of occurrence in NF2-SWN [7]. New molecular techniques allowed a more comprehensive routine mutation screening in these benign tumours and led to the detection of additional genetic events, such as variants in ERBB2 associated with mosaic SWN and responsive to lapatinib [8]. More recently a role for variants of KMT2 was uncovered in a rare intracerebral HNS [9]. The updated classification of SWN by Plotkin and co-workers in 2022 has shed new light on these tumours and their genetics [1].
Strictly speaking, the term ‘hybrid nerve sheath tumour’ is not precise. It is a collective term for different types of nerve sheath tumours that consist of components that are historically and conventionally assigned to one or another typical nerve sheath tumour. For SWN, only the HNS has a major role. With the introduction of the diagnosis of HNS into the different WHO classifications of tumours, this diagnosis should be made more frequently. But pathologists’ awareness is clearly limited. This is because (1) HNS is a benign tumour that is usually diagnosed without molecular analysis; (2) the variable proportion of the two different components can obscure one of them (which is why an Nf or an Sw is more likely to be diagnosed); and (3) the differential diagnosis may be difficult.
Take home messages
The schwannoma (Sw) and hybrid neurofibroma/schwannoma (HNS) are the characteristic peripheral nerve sheath tumours (PNST) in schwannomatosis (SWN).
HNS is a subtype of hybrid PNST that can easily be misdiagnosed.
Wrong classification of HNS as neurofibroma (Nf) can lead to misdiagnosis (instead of potentially associated SWN, the completely different neurofibromatosis type 1 might wrongly be suggested).
Conventional diagnosis of hybrid neurofibroma/schwannomaSchwannomas (Sw) are the most frequent tumours among benign PNST and are subdivided into conventional/classical, cellular, epithelioid, plexiform, and melanotic. The latter is typical for patients with Carney complex and PRKAR1A variants and not supposed to be typical in other hereditary conditions. Sw can be localised peripherally, viscerally, intraspinally, and intracranially. Sw can also be localised to very particular locations, as in the case of rare inner ear schwannomas (IES) [10]. Most IES cases appear to be sporadic, although a significant proportion are associated with NF2-related SWN [11, 12]. So far there are no reports of HNS in the inner ear. Given the phenotypic overlap between NF2- and SMARCB1/LZTR1-related SWN along with the potential contribution of mosaicism, further genetic studies are warranted in patients with IES [1, 13, 14].
The histological pattern of Sw comprises medium-sized, spindle-like tumour cells with long bipolar extensions and oval, elongated, or even roundish nuclei and arrangement in a fishlike pattern. Palisade positions of the tumour cell nuclei with alternating nucleus-rich and nucleus-poor being recognized as pathognomonic by Jose Verocay in 1910 and are referred to as Verocay bodies after him. The dense cellular pattern is referred to as Antoni type A (fibrillar type) after the Swedish physician Nils Ragnar Eugene Antoni. A tumour containing hypocellular, myxoid, or loosened accumulations of histiocyte-like, vacuolated, or fatty Schwann cells, is referred to as Antoni type B. A balanced ratio of Antoni type A and B areas is called conventional Sw. In the stromal part, thick-walled blood vessels with stores of hyaline material in the vessel walls are noticeable. Sometimes cavernoma-like vessels are formed. Fresh and older haemorrhages and abundant perivascular haemosiderophages are very typical. A schwannoma with regressive changes (so-called ‘ancient Sw’) demonstrates collagen deposits, fibrin thrombi, and pseudocystic regressions as well as a considerable nuclear pleomorphism and anisomorphism, which was formerly classified as a ‘degenerative polymorphic pattern’ and is not an anaplastic feature. A tumour with significantly increased cellularity and presence of a fascicular, storiform, or even non-characteristic picture is classified as a cellular Sw. Plexiform Sw offer a special growth pattern involving several nerve fascicles and intraneural growth. Despite various special forms, Sw are basically benign tumours that are classified as grade I according to the WHO classifications, although melanotic psammomatous Sw's are associated with a less favourable prognosis. Immunohistochemically, Sw shows an almost continuous positive immune reaction for the S100beta protein, with cytoplasm and nuclei being labelled. Furthermore, collagen IV marks the basement membranes of Schwann cells, which can also be visualised with silver (e.g., reticulin) staining. Experience has shown that the growth fraction, measured by the Ki67 staining index, is 2% to 5%, but can reach values of up to 15% in foci, particularly in cellular schwannomas. The loss of expression of LZTR1 and SMARCB1 may indicate SWN, although there is often only a partial loss, a ‘mosaic pattern’ [15]. Experienced neuropathologists cannot tell from histology alone whether a Sw occurs sporadically, singly, or multiply as part of a tumour syndrome or where it is localised in the body. The same applies to an Nf. However, if a plexiform structure is present or has been preserved by the operation, then a tumour syndrome can already be assumed, although there seem to be exceptions too (both plexiform Sw and Nf can appear to be sporadic although both may be due to very localised mosaicism) [15, 16].
Neurofibromas (Nf) originate from precursor cells of Schwann cells and are proliferations of both tumour cells (NF1−/−) sharing characteristics with unmyelinated Schwann cells and non-tumour Schwann cells (NF1+/−). They additionally contain nerve fibres, fibroblasts, macrophages, mast cells, and perineural cells interspersed with collagen, often within thick bundles. The Schwann cells display chromatin-dense wavy nuclei. Cytologic atypia or mitotic figures are typically absent in the benign tumour present in HNS. The tumour cells are embedded within a basophilic extracellular matrix enriched in glycosaminoglycans and collagen fibres. Plexiform growth is common in NF1. Immunohistochemically, partial immunoreactivity for S100 protein represents the proportion of Schwann cells. However, the number of S100 protein–positive cells is significantly lower compared with that in Sw. Additional focal immune-reactivities may be observed for glial fibrillary acidic protein (GFAP), vimentin, CD34, and factor XIIIa.
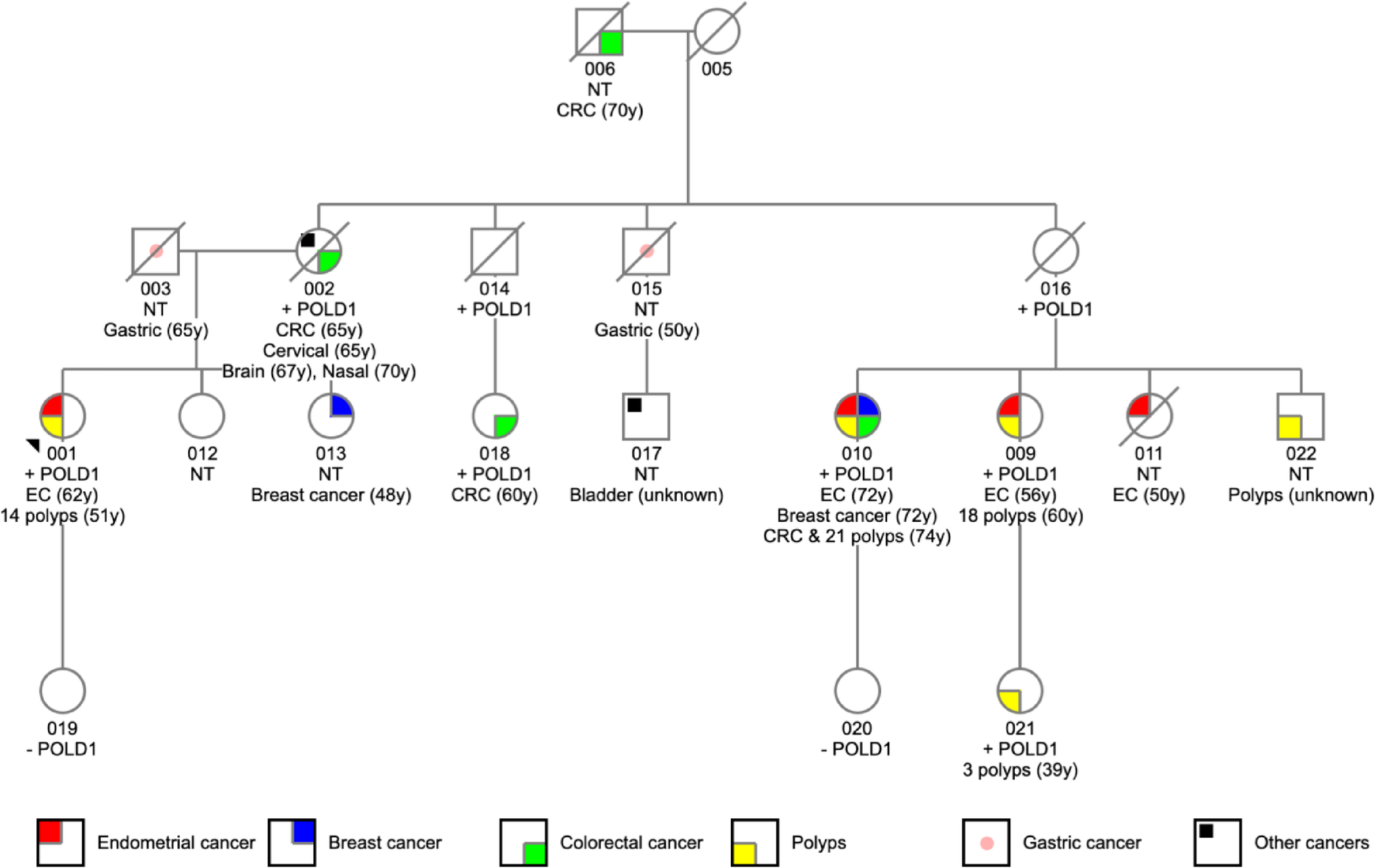
A hybrid peripheral nerve sheath tumour (HPNST) is a primarily benign neoplasm consisting of combinations of Nf, Sw, or perineuriomas (Pe) (Fig. 1). HPNST were first recognised in the WHO classification in 2013 [5, 6, 17]; the exact percentage of NST that are HPNST is not yet known. A recent review demonstrated that schwannoma/perineurioma is the most frequent type and is mainly sporadic [18, 19]. Although comprehensive studies are lacking, HNS are reported to constitute about 19% of HPNST [20]. Concerning HNS, to date only three larger studies have systematically analysed HNS tumour series, and all of them have demonstrated an association of HNS with tumour suppressor gene syndromes, especially SWN [2, 4, 7]. Salzano and co-workers have also recently reviewed some case reports [20]. Occurrence of HNS was rarely reported in NF1 (9%) but can be questioned looking back as these data were retrieved from submission pathology forms probably not all having been checked by geneticists; nevertheless, HNS might still occur in NF1, although they are increasingly reported in SWN [4]. In SWN, Sw and HNS occur at the identical locations involving nerves, mostly at peripheral or spinal nerves, but rare intracranial manifestations at the olfactory groove and intramuscular, intraorbital, and intraoral localizations are reported [9, 21,22,23].
Fig. 1
Benign hybrid peripheral nerve sheath tumours and their conventional tumours of origin. Although current studies document greater variability in the cellular tumour components and their origins, from a purely histological point of view schwannomas (Sw) are still supposed to share most characteristics with myelinating Schwann cells, neurofibromas (Nf) with non-myelinating Schwann cells, and perineuriomas (Pe) with perineural cells (indicated by the arrows originating from the appropriate structure of a normal nerve). The hybrid variants show histological characteristics of two different nerve sheath tumours and are here depicted between the classical entities (variant hybrid neurofibroma/schwannoma/perineurioma is not shown). SC, Schwann cells; TSC, tumour Schwann cells; L, lymphocytes; MC, mast cells; M, macrophages. As this review is mainly concerned with Sw, HNS, and Nf, typical histological patterns are shown (the photos are of tumours from previous studies [3, 4]). Created in BioRender. Leisz, S. (2025); agreement number: VH28JQMTK8; https://BioRender.com/iugbu4k
HNS consist of two histological intermingled growth patterns—those of typical Sw and of Nf, in varying percentages. Sw-like areas are mostly Antoni A regions; Antoni B regions may also occur, but only in large tumours. As typical for Nf, Nf-like areas often include abundant collagen deposits, myxoid changes, and fragments of neurofilament-positive, broken nerve structures. Nf-like areas are characterised by elongated and wavy-appearing tumour cells as seen in classical Nf. Alike in Sw and Nf, HNS can demonstrate a plexiform growth pattern, which was seen in nearly 30% of cases in one series [4]. In that series, it was also remarkable that Nf-like areas always surrounded the Sw-like nodules and that the Sw-like nodules were sharply encapsulated by collagen bundles from the surrounding Nf-like tissue. Many features typical of Sw are recapitulated in HNS, such as pronounced lymphocytic infiltrates. These Schwann cell–derived HNS express S100beta, which is dense in the schwannomatous component. The Ki-67 index is 0.8–18.5%. Until now, only one HNS has been described to show premalignant alterations with multiple chromosomal imbalances [3]. In a recent study, the Nf- and Sw-like HNS components were associated with distinct spatial gene expression clusters and transcriptional programmes [24]. Moreover, expression of APOD was proposed as a potential discriminator between these two components in seven follow-up cases. However, since the same study also demonstrated APOD expression in Antoni B areas of Sw and was based on only two tumours, further investigations meeting the criteria for a statistically robust power analysis are required to validate these findings. Nonetheless, this highlights the importance of distinguishing Antoni B areas from Nf-like regions of HNS in the diagnostic process.
To summarize, the required main criteria for a neuropathological diagnosis of an HNS include (1) occurrence of both Sw and Nf in the same lesion; (2) complete fulfilment by those areas typical of a Nf of the criteria for an Nf (grade I CNS according to the current WHO classification) for example, pronounced collagen deposition; and (3) complete fulfilment by areas typical of Sw of the criteria for Antoni A regions of an Sw (grade I of the current WHO CNS classification). On the basis of our experience, we suggest that at least 15% of the total HNS area should consist of either neurofibroma (Nf) or schwannoma (Sw) components to allow for reliable histological identification of both. Smaller proportions may be diagnostically challenging. In such cases, the tumour should be recut macroscopically and embedded more extensively to improve diagnostic accuracy. One should also check carefully that areas typical of Nf do not mimic Antoni B areas, by looking for evidence of collagen fibres and a mixture of Schwann cells and fibroblasts. One should be alert to tumours appearing with a plexiform growth pattern. Typical pronounced lymphocytic infiltrates and a sharp demarcation between the Nf and Sw components can be helpful minor diagnostic criteria.
SWN-related hybrid tumours are characterized by a typical mosaic staining pattern of protein SMARCB1 (positive and negative nuclei) and a remarkable loss of LZTR1 (only few positive nuclei left) in LZTR1-SWN. Involvement of other proteins of the chromatin remodelling complex has also been investigated [15]. Nevertheless, immunohistochemical staining—for example, for BAF170, NF2 protein merlin or others to show reduced protein expression due to LOH or mutations—might not be safe for routine diagnostic use. Therefore, the observation of important features from just daily routine haematoxylin and eosin (H&E)–stained slides (Fig. 2) is very helpful for the differential diagnosis. Advances in automated analyses based on deep learning could provide a cost-effective and rapid alternative to filtering HNS instead of performing expensive molecular analyses to ensure correct diagnosis.
Fig. 2
Typical histological pattern of hybrid neurofibroma/schwannoma (HNS). Right panel: Four different haematoxylin and eosin (H&E)–stained slides demonstrating the side-by-side combination of histological features characteristic of both neurofibroma (Nf) and schwannoma (Sw). Middle panel: CTNNA3 expression can be lower than normal in some HNS (compare upper picture with normal expression to picture below with reduced expression). In LZTR1-SWN, expression of the LZTR1 protein is lost (below) in nearly all nuclei. Left panel: In SMARCB1-SWN, a mosaic pattern of SMARCB1-positive and negative nuclei is detected. Photos are of tumours from previous studies [3, 4]. Created in BioRender. Lohse, S. (2025); agreement number: ZN28JQN57J; https://BioRender.com/a2zc7k7
Finally, new molecular single-cell analyses of Sw reveal that the cellular components are much more complex, with a greater diversity of Schwann cells than previously assumed: loss of the myelinating phenotype, myeloid cell infiltrations, so-called repair-type Schwann cells, and many more Schwann cell subtypes such as Schwann_VEGFA, Schwann_SCN7A, Schwann_PRX, and Schwann_CRLF1, according to their single-cell transcriptional and functional profiles [25, 26]. So far, however, this has not been applicable in achieving a histologically reliable and neuropathologically usable diagnosis.
Malignant transformation to MPNST arising from benign PNST in SWN has been described in a few studies so far even without history of radiation [27,28,29,30]. However, there is little reliable knowledge about the processes of malignant transformation in HNS. However, individual descriptions indicate that further molecular changes associated with malignancy may also occur in the HNS [3], meaning that accurate histological diagnosis remains an important tool in everyday routine practice for identifying underlying abnormal patterns and referring them for molecular analysis.
Take home messages
Diagnosis of an HNS requires occurrence of both Sw-like and Nf-like tumour areas in the same lesion that, respectively, fulfil the criteria for an Nf and an Sw (grade I CNS WHO classification).
A plexiform growth pattern is common, but every plexiform peripheral nerve sheath tumour should suggest the presence of a tumour syndrome and requires special diagnostic care.
HNS often show pronounced lymphocytic infiltrates and a sharp demarcation between the Nf and Sw components.
Molecular diagnosis of a hybrid neurofibroma/schwannomaSince the correct diagnosis of HNS can be extremely important for the differentiation of NF1 and SWN, molecular diagnostics may be of particular importance in special cases. This was demonstrated in our daily practice in a case in which there were enquiries from the clinical side because the past diagnosis of a Nf did not match the clinical picture in any way. A detailed family history, clinical examination and genetic diagnostics revealed a mutation in the LZTR1 gene, which led to the re-diagnosis of all the patient's tumours, which turned out to be exclusively HNS at different locations. Nevertheless, it turns out that the number of molecular analyses of HNS at the somatic level is very limited. This may be due to the fact that these tumours are still too rarely diagnosed, and thus no larger series are created that are suitable for high-throughput analyses. Until the contrary is proven, the only question that remains is whether HNS, although they are distinguishable from Sw by histology, represent a separate molecular entity, because they show the molecular changes we see in Sw. The question remains if they are only a morphologic variant of Sw.
The fact that HNS might be a variant of Sw is supported by data from Röhrich et al. [31], who reported that tumour methylation profiles of HNS cluster with those of benign Sw. Sw were described to form four different methylation subgroups (I–IV), and HNS were mostly localized in cluster II. In support of this, two HNS investigated by spatial gene expression analysis were also found to cluster to the Sw methylation class [24]. However, other methylation clusters have been described by Agnithori et al. for Sw [32].
Since only a few molecular data have been collected for HNS, but a great deal for Sw, we first look at Sw, especially since HNS may represent only a subtype of Sw at the molecular level. For the development of Sw in LZTR1- and SMARCB1-related SWN, a multistep-model involving at least four hits or three steps has been postulated [33, 34]. Knudson’s two-hit model needed to be refined due to a higher complexity concerning genes involved in SWN such as LZTR1, NF2, and SMARCB1, and rarely SMARCCA4 and COQ6 [35, 36]. A typical and frequent somatic (second) hit (about 61% in all Sw according to Agnithori and co-workers) in Sw is complete loss of chromosome 22 or of 22q, including all or some of these genes located on chromosome 22 (reviewed in detail by Kehrer‑Sawatzki et al.); thus, indicating that at least two different tumour suppressor genes are involved [32, 34]. Beyond LZTR1, NF2, SMARCB1, SMARCA4, COQ6, more genes have been detected to be involved in Sw pathogenesis. These include genes encoding the chromatin modifiers ARID1A/B, DDR1, TSC1/2, and a recurrent SH3PXD2A:HTRA1 fusion (balanced translocation on 10q) and some other rare [32, 37]. Interestingly, CDKN2A germline variants have been detected in SWN patients with melanoma as well as PNST that seem to match HNS, and DGCR8 variants have been detected in SWN patients with euthyroid multinodular goiter [37,38,39]. Albeit not in this abundance, HNS have been demonstrated to show involvement of nearly the same set of genes and frequent monosomy 22 [3].
In general, genetic variants in HNS comprise germline and somatic events that are also detected in Sw in genes such as NF2, LZTR1, and SMARCB1. These genetic variants need to be differentiated and might help to define a genetic disease such as SWN if the germline mutations are confirmed (e.g., from blood cells). Detection of pure somatic pathogenic variants indicates mosaicism or postzygotic events that occur after the fusion of the male and female germ cells to form the zygote, as shown for somatic variants of ERBB2, RET, and KMT2A and for a rare gene fusion RREB1::LPP [8, 9, 19]. It is not known if specific somatic variants are associated with specific tumour localization; one might suspect this of specific manifestations such as the rare intracranial HNS occurrence described in a single report [9]. The recent study by Goto and co-authors identified a somatic KMT2A variant, c.4408C > T, p.Q1470*, and trisomies (chromosomes 5 and 14q) while other gene variants (NF1, NF2, chr. 22 loss), especially germline mutations, were excluded. Furthermore, in one of 22 cases, Stahn et al. reported a focal constitutional deletion of CTNNA3 located at chromosome 10q21.3 [3]. In addition, transient knockdown of CTNNA3 in Schwann cells resulted in cytoskeletal alterations and reduced E-cadherin expression, indicating aberrations of epithelial–mesenchymal transition (EMT)-like processes. The utmost recurrent event is a heterozygous deletion of chromosome 22 (monosomy) or at least 44% of 22q that varies within the studies. However, to date, besides a limited transcriptomic approach of two cases [24], the molecular characteristics of HNS have been investigated only in very small case series by Harder et al., Goto et al., and Ronellenfitsch et al. [4, 8, 9], whose main findings are summarized in Table 1 and Fig. 3.
Table 1 Summary of genetic alterations in SWN-associated HNS (NF2-related or non-NF2-related)Fig. 3
Overview of pathogenic variants in hybrid neurofibroma/schwannoma (HNS)
Comments (0)