Chemicals, supplies, and samples
The analytical standards for the target analytes, namely 4:2 FTOH (1H,1H,2H,2H-Perfluorohexan-1-ol), 6:2 FTOH (1H,1H,2H,2H-Perfluorooctan-1-ol), 8:2 FTOH (1H,1H,2H,2H-Perfluorodecan-1-ol), 10:2 FTOH (1H,1H,2H,2H-Perfluorododecan-1-ol), 8:2 FTAc (1H,1H,2H,2H-Perfluorodecyl acrylate), N-MeFOSA (N-Methylperfluorooctanesulfonamide), N-EtFOSA (N-Ethylperfluorooctanesulfonamide), N-MeFOSE (N-Methylperfluorooctanesulfonamidoethanol), and N-EtFOSE (N-Ethylperfluorooctanesulfonamidoethanol) were sourced from LGC Limited (Teddington, Middlesex, UK). Ultrapure water was produced using a Milli-Q system from Millipore (Bedford, MA, USA), and the C7-C30 linear alkane mix was obtained from Supelco (Bellefonte, PA, USA).
Stock standard solutions of each analyte (1000 mg L⁻1) were prepared in MS grade methanol (Supelco) and stored at −20 °C in the dark. Working solutions were prepared by diluting the stock solutions in ultrapure water to various concentrations.
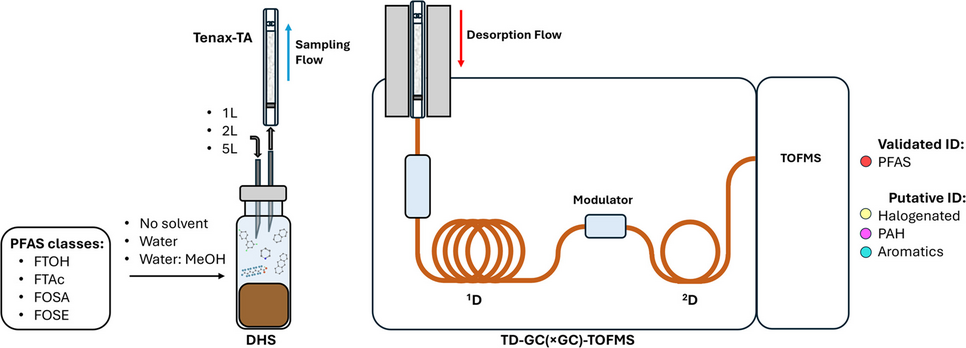
Thermal desorption tubes (TDT) containing Tenax-TA (TA) as a sorbent (Supelco) were used for the DHS extractions. The desorption tubes were conditioned after each extraction and analysis cycle using a Phoenix T220 (REDshift s.r.l., San Giorgio in Bosco, Padua, Italy) with a ramp rate of 30 °C min⁻1 to 320 °C, held for 180 min, under a nitrogen flow of 100 mL min⁻1. Tube blank analyses were periodically conducted to prevent carryover.
Dynamic headspace extraction
To evaluate the influence of different auxiliary solvents on PFAS recovery, three different conditions were examined: soil with no solvent addition, soil with added pure water, and soil with a 1:1 solution of pure water and methanol. Nine airtight headspace glass vials (20 mL capacity) were prepared, with three replicates for each condition. Firstly, 2.00 g of soil were added into the vials and spiked with the PFAS standard solution to achieve a concentration of 10 ng g−1. The vials were left overnight to allow the compounds to absorb into the soil matrix. Before extraction, nine spiked vials were divided into three groups (n = 3 each). Five milliliters of pure water were added to the vials of the first group, and 5 mL of the 1:1 water to methanol solution were added to the vials of the second group, while no solvent was added to the vials of the third group. Each vial was conditioned for 15 min at 30 °C with magnetic stirring (~ 800 rpm) and then connected to the sampling apparatus consisting of a pre-conditioned tube and a vacuum pump, which maintained an 80 mL min−1 sampling flow rate, for a total extraction volume of 1 L (ca. 12.5 min sampling time). For this research, Tenax-TA (a porous polymer of 2,6-diphenyl-p-phenylene oxide) was used as the sorbent material for its superior performance on the selected analytes, as previously demonstrated [20]. The pump was calibrated and the flow monitored with a flow meter during each sampling. After the extraction step, each tube was desorbed into the GC-TOFMS system and analyzed. Tukey’s test at a 95% confidence level was performed to compare the mean yields of each PFAS in the three different extraction conditions; the same approach was also applied to the total PFAS yield.
Regarding the extraction volume, three different volumes were evaluated (1 L, 2 L, and 5 L). Similarly to the solvent assessment, nine vials (three replicates for each volume) were spiked at a concentration of 10 ppb and equilibrated overnight. After matrix homogenization, 5 mL of a 1:1 solution of pure water and methanol were added, and the samples were extracted.
The real soil sample was also extracted using the selected volume under the same conditions and analyzed by GC(×GC)-TOFMS. Tukey’s test at a 95% confidence level was performed to compare the results.
Instrumental experimental conditions
Both GC-MS and GC×GC-MS analyses were carried out using an Agilent 8890 GC coupled to a Pegasus BT 4D time-of-flight mass spectrometer (LECO Corporation, Mönchengladbach, Germany). A PAL System automated tube handling device (CTC Analytics AG, Zwingen, Switzerland) was used to place the tubes inside an Optic-4 multimode injector automatically (GL Sciences B.V., Eindhoven, Netherlands), featuring an inlet Peltier cooler and a liner exchanger (GL Sciences B.V.). The GC system also included a cryogenic trap (GL Sciences B.V.) for the secondary trapping and release stage. Chromatographic separation was achieved using an Rxi-5ms column (30 m × 0.25 mm × 0.25 μm df) for the first dimension and an Rxi-17SilMS column (2 m × 0.25 mm × 0.25 μm df) for the second dimension (both from Restek Corporation, Bellefonte, PA, USA). Helium (99.9999% purity) was used as the carrier gas.
After sampling, the TDTs were removed from the sampling device, capped, and placed in the PAL autosampler. Once placed in the thermal desorption inlet, a 4-min solvent venting step was applied (split flow of 20 mL min−1 and a column flow of 0.5 mL min−1, at 35 °C) and followed by the thermal desorption step, increasing the temperature from 35 °C to 300 °C with a quick ramp of 20 °C s−1, maintaining a split flow of 20 mL min−1 and a column flow of 1.5 mL min−1. The cryogenic trap temperature was maintained at −20 °C throughout the venting period (4 min) and the thermal desorption phase (45 s) to facilitate analyte trapping. Its temperature was then quickly raised to 300 °C at a rate of 50 °C s−1 (held for 3 min), allowing for the secondary release of the analytes.
The initial temperature of the GC oven was set at 40 °C (held for 1 min), then increased to 175 °C at a rate of 5 °C min−1. After reaching 175 °C, a rapid ramp of 30 °C min−1 was performed to reach the final temperature of 300 °C (held for 1 min), resulting in a run time of 33.10 min. The secondary oven and modulator temperature offsets were respectively set at + 10 °C and + 25 °C compared to the main oven. Regarding the GC×GC analysis, the oven temperature was set at 40 °C (held 1 min), then increased at 5 °C min-1 to 300 °C (held for 1 min), and the modulation period was 3 s (hot jet: 0.8 s; cold jet: 0.7 s). ; in 1D-GC analysis, the modulator was simply turned off. The MS transfer line temperature was set at 280 °C, and the electron ionization ion source temperature was set at 250 °C, with an ionization energy of 70 eV. The TOF was operated across a mass range of 29 to 600 m/z, using an acquisition delay of 180 s at a frequency of 10 Hz and 150 Hz for one-dimensional and two-dimensional analysis, respectively. Data acquisition and processing were performed using ChromaTOF software version 5.56 (LECO Corporation).
The quantification ion for each analyte was selected based on the mass providing the best selectivity and detectability, with a tolerance of 500 mDa. The quantifier ions are reported in Table 1.
For peak detection in non-targeted analysis, a signal-to-noise (S/N) of 10 was set. Target analytes were identified using standards (Validated ID). Putative identifications were based on mass spectral similarity and chromatographic features compared with the NIST23 library. Two levels of reliability were applied for the tentative identification of environmentally relevant analytes (Figure S4): a more reliable ID level, Putative IDi, was based on a combination of spectral (i.e., MS similarity > 85%) and chromatographic information (i.e., RI tolerance ± 20, position on the 2D plot), while a weaker level of identification (Putative IDii) was based only on the match of the MS fragmentation with reference databases (MS similarity > 75%) and the two-dimensional structured elution. The retention indices were calculated using a C7 to C30 alkane series and analyzed under the same conditions as the samples.
Blank system (with no sample tube desorption) was periodically run to exclude any carryover.
Calibration and method validation
A five-level calibration curve was built for each of the nine PFAS using the previously described optimized conditions: 2.00 g of soil were added in the headspace vial and spiked with the mix of the nine PFAS standards in order to reach different final concentrations (10, 20, 30, 40, and 50 ng g⁻1). Three replicates were prepared for each level and left overnight for equilibration; then, 5 mL of a 1:1 mixture of pure water and methanol were added before extraction. The solutions were extracted using 1L volume under agitation (approximately 800 rpm), and the tubes were analyzed by GC-TOFMS. To validate the method linearity, accuracy (calculated using 25 ng g⁻1 as the percent difference between the expected and measured concentrations), precision (expressed as the relative standard deviation (RSD%) at the lowest level of the calibration curve), limit of detection (LOD), and limit of quantitation (LOQ) were calculated.
Even though an internal standard is generally helpful to monitor method variability, none was used in this study. However, our recent study showed very reproducible results on IS response [20] using the same extraction conditions for volatile PFAS.

Comments (0)