
Remember me
All reported numerical values are presented as mean (standard deviation) of replicate measurements, with standard deviations (SD) indicated in parentheses. When appropriate, relative standard deviations (RSDs, %) are also specified.
MaterialsUltrapure water obtained from a Milli-Q® IQ 7000 purification system (Merck Millipore, Germany), equipped with a 200-nm polyethersulfone filter to ensure particulate and bacteria-free water, was used for the dissolution and filtration of the tablet matrix and for cleaning procedures. PP pellets (Moplen RP320M, LyondellBasell) were used to prepare PP powder RMs. Directly compressible lactose (FlowLac90, D50, 138.2 (3.1) µm) was provided by MEGGLE GmbH & Co. KG. and PEG 6000 (D50, 98.5 (2.2) µm) was purchased from Merck. Ethanol absolute (99.9%, CHEMSOLUTE) was also used for various cleaning procedures.
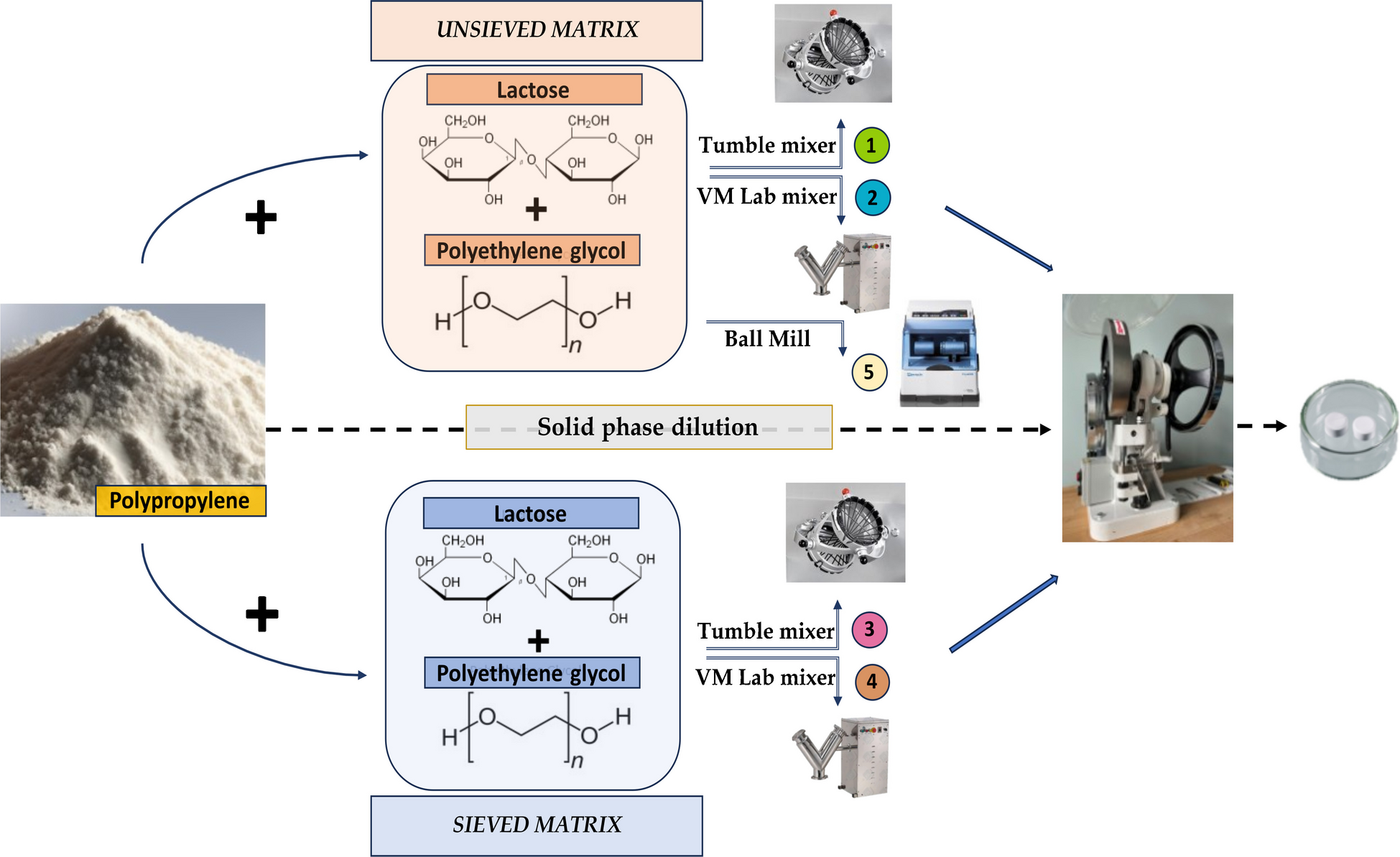
Production of reference material tablets containing PP microplasticThe production of RM tablets containing a PP MP involved a multi-step process that included preparation of the PP MP from pellets, mixing of the MP and matrix components, and compression into tablets. Considering the two variables, (i) the particle size distribution of the matrix and (ii) the homogenization mixer instrument, a set of five different tablet samples was prepared by pressing the PP MP RM with the size range 1–100 μm in a water-soluble matrix composed of lactose and PEG 6000. The PP MP was combined with either sieved or unsieved carrier matrix using three different mixer instruments, allowing the homogeneity and stability of the resulting RMs to be evaluated according to these parameters. The homogenization mixer instruments employed for tablet preparation included a Tumble mixer (LM-TM100, Laarmann, Roermond, Netherlands), VM Lab mixer (LFA machines), and Ball Mill (Retsch® CryoMill 100–240 V, 50/60 Hz). The preparation conditions for each sample are summarized in Table 1, and the mixing instruments used are shown in Figure S1.
Table 1 Preparation conditions for PP tabletsPreparation of PP microplastic and matrix componentsMP particles were obtained by cryomilling of PP pellets using an Ultra Centrifugal Mill ZM200 (Retsch, Haan, Germany) with a rotation speed of 16,000 rpm. The mill was equipped with a cyclone. The cryogenic grinding of the pellets was conducted at low temperature using liquid nitrogen. A ring sieve with a 500-µm mesh size and a 12-teeth rotor was selected for the procedure. Before starting the milling process, the system was pre-cooled by adding approximately 60 mL of liquid nitrogen directly into the mill chamber. Meanwhile, the PP pellets were placed in a separate container and cooled with liquid nitrogen for a few minutes before being slowly introduced into the mill. The feeding process was carried out by alternating a maximum of five pellets, kept immersed in liquid nitrogen, with approximately 60 mL of only liquid nitrogen. The continuous addition of liquid nitrogen ensures that the rotor remains at a low temperature and prevents overheating, while limiting the number of pellets helps avoid the formation of aggregates that could interfere with the rotor movement. After the milling process, the resulting powder was transferred into a glass beaker, covered with aluminum foil to prevent contamination, and placed in the vacuum drying cabinet for at least 2 h (25 °C, 15 mbar).
To isolate MP particles in the 1–100 μm size range, the dried powder was fractionated with a dry-sieving method using a Retsch™ sieve shaker AS 200 Control (Retsch, Haan, Germany). Stacked stainless-steel sieves (20 cm in diameter) with mesh sizes of 500, 100, and 50 μm were shaken for 20 min at an amplitude of 1.40 mm and an interval time of 5 s. For each sieving cycle, the powder volume was set to 88 cm3, corresponding to the maximum capacity of the sieves. This volume was measured by carefully filling a graduated cylinder with 88 mL of powder, which is equivalent to 88 cm3 (1 mL = 1 cm3). The mass of powder for each cycle was then determined based on the measured volume and the density of each material, resulting in approximately 40 g for cryomilled PP powder, 60 g for lactose, and 54 g for PEG 6000. Minor variations in powder packing may occur during measurement, which could slightly affect the actual mass per cycle. Lactose (D50, 138.2 (3.1) µm) and PEG 6000 (D50, 98.5 (2.2) µm) powders were utilized as received from the supplier to produce three sets of tablets, while a separate batch of each matrix component was also sieved prior to use to isolate particles of a similar size range to the PP powder (1–100 µm). The sieving conditions for the matrix components were analogous to those applied to the PP. Due to differences in density, the maximum amount of material used in each sieving cycle was 60 g for lactose and 54 g for PEG 6000. Particles in the 1–100 µm size range were obtained by combining the yields from the 100–50 µm and < 50 µm mesh size. Additional information about the sieving process is available in the developed standard operating procedure (SOP) [22].
Mixing and compression into tabletsTablets were produced by solid-phase dilution and the subsequent pressing of 200 tablets of each sample type (Table 1). The solid-phase dilutions were prepared with PP in the size range of 1–100 μm, which was mixed with lactose and PEG 6000 as the matrix components. Depending on the specific sample type, two different matrix conditions were applied: (i) lactose and PEG 6000 were used as received (unsieved) or (ii) lactose and PEG 6000 were sieved to the same size as the PP. Three different homogenization methods were then applied to the solid-phase dilution to assess their impact on the resulting PP content of the tablets: Tumble mixer, VM Lab mixer, and Ball Mill (Figure S1). Each combination of matrix condition and mixing instrument defined a specific sample, as previously listed.
Each individual tablet component was added into the homogenization mixer instrument and thoroughly mixed between different dilution steps to ensure homogeneous distribution of PP particles in the individual tablets. The sample preparation procedure for the Tumble mixer and the VM Lab mixer, which comprised 5 rounds of dilution and mixing, is summarized in Table S1. In the case of the VM Lab mixer, the mixing time for each step had to be extended, as the number of rotations of the mixing vessel was insufficient to ensure proper homogenization at the same time required for the Tumble mixer. For sample 5 (unsieved matrix, Ball Mill), the procedure was adapted to the specific requirements of the mixing instrument. Homogenization was carried out under liquid nitrogen, with a pre-cooling time of 10 min, six mixing cycles of 2 min each, and 2 min of cooling down during the individual cycles. A 50-mL grinding vessel with a single large and heavy ball of 25-mm diameter was used. The vessel was filled with 1/6 grinding ball, 1/6 sample, and 2/3 remaining free space, the latter required for ensuring ball movement and to account for possible sample expansion. Due to the limited volume of the grinding vessel, only 10 g of the solid-phase dilution could be used. Therefore, the quantities of the PP powder and matrix components were recalculated proportionally to match the 10 g target and directly added to the vessel to start the homogenization process. The sample preparation for Ball Mill is summarized in Table S2.
After each individual sample mixture (Table 1) had been produced, it was stored overnight in a vacuum drying cabinet (50 °C, 30 mbar) to remove any residual water prior to the tableting process. Tableting was performed with a TDP 5 Desktop Tablet Press (LFA Machines Oxford LTD, UK) operated in mode to ensure that the individual tablets were visually identical and had no breaking edges. To prevent the tablets from flaking, the stamp was cleaned at regular intervals with ultrapure water and ethanol, before being dried. Control weighing and solubility assessment were conducted in 10% of the tablets to ensure a constant tablet mass of 250 (5.0) mg and complete dissolution in water.
A comprehensive overview of the preparation of tablets is presented in Fig. 1.
The prepared PP RM tablets were subsequently analyzed using different analytical techniques by different laboratories and operators, each applying its own sample preparation workflows for the different techniques according to availability and the PP MP particle size fraction.
Fig. 1
Production process for the PP microplastic reference materials
Characterization of tablets and their individual componentsParticle size measurement by laser diffractionParticle size distribution measurements of the dry PP powder, lactose, and PEG 6000 (sieved and unsieved) were performed by LD using a HELOS BR instrument (Sympatec GmbH, Clausthal-Zellerfeld, Germany). The RODOS dry dispersion system with an injector width of 4 mm and a pre-pressure of 2.5 bar in combination with the micro dosing unit ASPIROS (transportation sleigh speed 100 mm/s) was applied. From 30 to 50 mg of powder was placed into a device-specific glass vial for analysis. The measurement time was 10 s, and 3 replicates were made with an R5 (4.5–875 µm) or R3 (0.9–175 µm) lens, according to the expected particle size range. All measurements were processed with the PAQXOS 4.1 software (Sympatec GmbH). Prior to the particle size measurements, the PP powder was weighed and passed through a deionizing device (Haug EN SL LC Generator combined with PRX U Frame Set, HAUG GmbH & Co. KG, Germany) to neutralize electrostatic charges on the particle surfaces. This treatment reduces agglomeration and improves the uniformity of dry dispersion within the laser diffraction system, thereby minimizing potential electrostatic effects on the measurement. Together with the use of three replicate analyses, this procedure contributed to obtaining consistent and reproducible particle size distributions, including the low size range.
ATR-FTIRFTIR spectroscopy was performed using a Thermo Scientific™ Nicolet 8700 FTIR spectrometer in ATR mode to confirm the polymer composition of PP powder, ensure that no chemical or structural changes occurred during the production and sieving process, and guarantee unambiguous identification using other spectroscopic methods. The spectra were recorded from 500 to 4000 cm−1 with 32 scans and a resolution of 4 cm−1. The data obtained were processed with Omnic software, identifying the substances according to the spectral database.
Raman spectroscopyMicro-Raman analysis was performed on PP MP particles collected on silicon (Si) filters after dissolving the PP tablets in water and subsequent filtration, using a LabRAM HR Evolution spectrometer (HORIBA France SAS). Each tablet was processed on different days by dissolving it directly in a glass funnel containing 1 L of reference water, previously analyzed and validated as free from MPs. At the end of the filtration, a solution of Triton™ X-100 (0.1% v/v in water, Thermo Fisher Scientific) was added to reduce particle agglomeration. Filtration was performed using Si filters with a pore size of 5 µm (macroporous silicon membrane, Smart membrane) to ensure effective retention of the majority of PP MP particles (1–100 µm) while maintaining an acceptable filtration flow rate. The filters were subsequently stored in closed Petri dishes until Raman analysis.
For each series of filtrations, a procedural blank was done with reference water prior to the filtration of PP tablets to evaluate the MP pollution that could occur during the filtration process.
All the manipulations were performed under a laminar flow hood. The laboratory adhered to the precautions outlined in [23], including hand washing, refraining from wearing makeup, and using cotton lab coats.
Spectra were acquired in the range of 550–2000 cm⁻1 using a 532-nm excitation laser with a power of 100 mW, an acquisition time of 0.1 s, and one accumulation. A 50× objective was employed for all measurements. Data acquisition was carried out with LabSpec software (v. 6.8.1.9), and particle identification was conducted using IDFinder software (v. 4.2, HORIBA France SAS). Spectra with a Hit Quality Index (HQI) > 70% were automatically assigned to PP, while those with 40% < HQI < 70% were manually verified.
Scanning electron microscopySEM was used to investigate the shape, surface morphology, and size of the particles. The PP, as well as sieved and unsieved lactose and PEG 6000 powder, was placed on double-sided adhesive carbon tape on the aluminum SEM stubs and coated with a 10-nm conductive gold layer to achieve surface electrical conductivity. High-resolution surface imaging of the particles was obtained using an EVO MA 10 SEM (Carl Zeiss Microscopy GmbH, Oberkochen, Germany) operated at an acceleration voltage of 10.00 kV and equipped with a tungsten cathode and a conventional secondary electron detector. Micrographs were captured at a resolution of 1024 × 768 pixels, a magnification range of 500×, and a working distance of 7 mm.
Thermogravimetric analysisTen tablets of each sample type were analyzed by TGA (Metter-Toledo TGA/DSC 3+) to assess the homogeneity and stability of the PP tablets. Samples were filtered through a specialized smart filter crucible (stainless-steel microfilter crucibles, 5 μm pore size, 9 mm in diameter, and 13 mm in height) (Gebr. Kufferath AG, GKD) [24] using approximately 50 mL of ultrapure water to dissolve and remove the tablet matrix. This innovative crucible design served a dual purpose: it functioned as a filtration device for collecting the PP MP particles from the dissolved tablet matrix and, critically, as an inert and thermally stable sample holder for subsequent thermal analyses, including both TGA and the hyphenated TED-GC/MS described later. After the filtration, the collected particles were stored in the oven at 50 °C for at least 3 h and later in the desiccator for another 2 h. Dried crucibles were weighed before and after filtration to calculate the mass of each sample. The dried loaded crucibles were placed in the TGA autosampler and heated from 25 to 600 °C [25] at 10 K/min under an inert atmosphere (N2, 30 mL). The decomposition temperature of polymers typically occurs between 350 and 500 °C [26]. The individual tablet components were also measured to determine their decomposition temperature range by measuring up to 1 mg of PP, lactose, or PEG 6000 separately in crucibles three times. The corresponding thermogravimetric mass loss and the corresponding first derivative curves were analyzed. Stability control was performed after 4 months on 7 samples.
Thermal extraction desorption-gas chromatography/mass spectrometryTen tablets from sample 3 were each analyzed by TED-GC/MS, which combines TGA (Mettler Toledo TGA 2 Star) with gas chromatography-mass spectrometry (GC/MS) (7890 B and 5977 B MSD, Agilent Technologies, Santa Clara, USA) [27]. Each tablet was first filtered through the smart filter crucible (as described in section “Thermogravimetric analysis”) and the collected PP MP particles in the dried crucibles are decomposed by TGA under a N2 atmosphere (from 25 to 600 °C), and the gaseous decomposition products are captured on a solid-phase sorbent, poly(dimethylsiloxane) (PDMS) (Sorb‑Star, ENVEA GmbH), for subsequent GC/MS analysis. A thermal desorption step was conducted by heating each sorbent to 200 °C, with the analytes subsequently cryo-focused on the cold injection system at −100 °C using liquid nitrogen. The decomposition products were then injected and separated by the GC column and analyzed in the MS. Additional information about a typical TED-GC/MS analysis is available in Nature Protocol [28]. PP was quantified using 2,4,6,8-tetramethylundec-10-ene as a marker (m/z 111).
Pyrolysis gas chromatography-mass spectrometryTen tablets from sample 3 were each analyzed by Py-GC/MS. PP particles from the tablets were isolated by filtration using a stainless-steel filter holder with a Whatman glass fiber filter (1.6 µm pore size, 13 mm filter size), following several validated protocols [29], and a 20-mL glass syringe connected to a VisiprepTM vacuum manifold (Supelco Sigma-Aldrich Co.). PP tablets were placed in the syringe barrel and half-filled with ultrapure water. After 4 min to allow dissolution, the sample was rinsed several times with additional ultrapure water, for a total volume of 150 mL, to ensure complete matrix removal. The filter was folded and placed in the pyrolysis ECO-cup (80 µL, Frontier Labs) and dried in an oven at 40 °C for a minimum of 4 h. Analysis was performed using a Frontier Multi-Shot Pyrolyzer (PY-3030D) (Frontier Labs, Japan) coupled with an Agilent 8860 GC and an Agilent 5977 A MSD (Py-GC/MS) (Santa Clara, CA, USA). The pyrolizer operated in single-shot mode with pyrolysis at 600 °C for 0.5 min. The pyrolizer interface and GC inlet temperatures were set to 300 °C, and the split ratio was 100:1. Helium was used as the carrier gas at a constant flow rate of 1 mL/min. Separation was achieved using an HP-5MS UI capillary column (30 m length, 0.25 μm film thickness, and 0.25 mm internal diameter). The column oven temperature was programmed to start at 40 °C for 2 min, then increased at a rate of 20°C/min up to 325 °C (14 min), followed by an increase of 30 °C up to 340°C. The transfer line temperature was maintained at 320 °C, the ion source temperature at 280 °C, and the quadrupole temperature at 150 °C. The ion source operated in full scan mode. PP was quantified using 2,4-dimethylhept-1-ene as a marker (m/z 70, 126) and 2,4,6,8-tetramethylundec-10-ene as a marker (m/z 69, 111) as confirmation markers, using external calibration.
Comments (0)