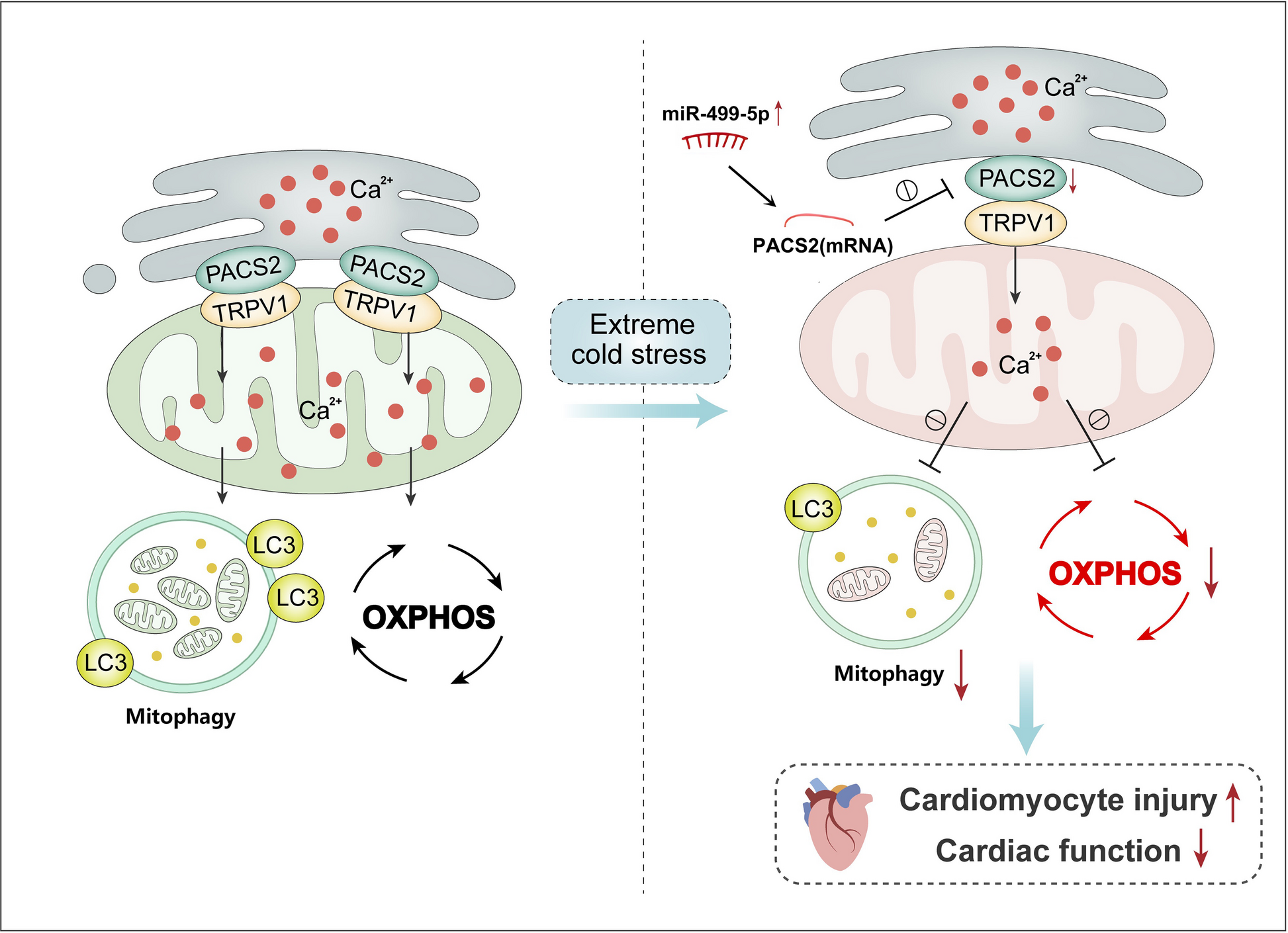
Mouse Model
SPF Biotechnology supplied 6-week-old male C57BL/6N mice, which were placed in a controlled thermal chamber for 3 days without any temperature intervention, to allow for environmental acclimatization. They were provided with unlimited access to a standard diet (SPF-F01-001, SPF Biotechnology Co., Ltd.) and water, and housed under an automatically controlled 12-h light/dark cycle [17]. The mice were divided into two equal groups based on their body weight (22.14 ± 0.26 vs. 22.15 ± 0.25, p = 0.973): a control group maintained at an average temperature of 22℃, and a treatment group that was exposed to −20°C for 4 continuous hours daily over a period of 2 weeks. Cold exposure began 2 h after the light was turned on.
Echocardiographic Measurement
Mice were anesthetized using 1.5% isoflurane, and their cardiac geometry and function were evaluated through echocardiography using the Vevo 3100 system (VisualSonics, Toronto, Canada), which was equipped with a 21-MHz microprobe. LV function was measured using M-mode and Doppler the day before the sacrifice. The average of three consecutive cardiac cycles was calculated for each parameter and the echocardiographic images were blindly analyzed by two independent researchers.
Histological Analysis
The posterior LV wall was dissected and preserved in 4% paraformaldehyde. It was then embedded in paraffin and sectioned into 5 μm slices. These sections were stained with Hematoxylin & Eosin (HE) (C0105S, Beyotime) as well as Masson’s trichrome staining (C0189S, Beyotime). Light microscopy was used to observe the stained sections. To analyze the cross-sectional area (CSA), the heart sections were stained with Wheat Germ Agglutinin (WGA) (W11261, Invitrogen). A minimum of three cross-sections from each heart were examined and averaged for statistical purposes. Quantification of the areas was carried out using the software of ImageJ V1.8.0.112.
Biochemical Analysis
The levels of natriuretic peptide (BNP), troponin I (TnI), and creatine kinase-MB (CK-MB) in mouse plasma were quantified using the Enzyme Linked Immunosorbent Assay (ELISA) method, with duplicate measurements performed using a commercial mouse kit provided by Jingmei Biological Technology Company. These measurements were conducted strictly following the manufacturer’s guidelines. Approximately 2mL of blood was drawn from each mouse and stored in a tube containing purified hemagglutinin and organic silicon active substances for coagulation. Subsequently, the blood was allowed to clot at room temperature for 10 min and then centrifuged at 3000 g for 20 min.
Cardiomyocyte Isolation
Adult mouse LV cardiomyocytes were isolated using the Langendorff system and collagenase digestion [15]. Briefly, mice were anesthetized with isoflurane, injected with cefoperazone (2mg/kg) and heparin (10U/g) abdominally, and then their hearts were quickly removed and rinsed with 0.9% normal saline. The hearts were cannulated to the 18G cannula via the aorta and perfused with bicarbonate buffer [containing 120mM NaCl, 5.4mM KCl, 1.2mM MgCl2, 0.3mM Na₂HPO₄, 1 mM 2,3-butanedione monoxime (BDM), 20mM 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES), 10mM taurine, 10mM glucose, and 2mM NaOH, pH 7.4] for 5 min. The perfusion rate was approximately 4-5mL/min. Enzymatic digestion was initiated by perfusing the heart with digestion buffer (containing 50 ml perfusion buffer, 1.4mg/mL collagenase II, 0.6mg/mL protease XIV and CaCl2 50μM) and recirculating for 12–15 min. All solutions were maintained at a constant temperature of 37℃ and continuously bubbled with 95% O₂/5% CO₂. Then the ventricles were removed, mechanically dissociated, and filtered through a 140nm nylon mesh. The filtered tissue was transferred to a bicarbonate buffer containing CaCl2 and the Ca2+ concentration was adjusted to 500uM using a stepwise calcium compounding method at room temperature (with sequential 10-min incubations at 100μM, 200μM, and 500μM CaCl2). The suspension was placed in a 50mL conical tube for 10 min and then cultured overnight in Eagle medium. Before the experiment, the culture medium was replaced, and unattached cells were removed.
Mitochondria Isolation
The cardiomyocytes were collected and resuspended in an ice-cold buffer [225mM mannitol, 75mM sucrose, 0.1mM Ethylene Glycol Tetraacetic Acid (EGTA), and 30mM Tris–HCl pH 7.4]. Then, the cells were homogenized using a Dounce homogenizer, and cell integrity was checked under a microscope. Homogenization was completed when 80–90% of the cells showed damage. After that, the cell extracts were centrifuged twice at 600 g for 5 min at 4℃. The supernatant was then centrifuged three times at 8,000g for 10 min at 4℃. The pellet, containing the pure mitochondrial fraction, was collected by discarding the remaining supernatant. This crude mitochondrial pellet was layered in 2 ml of ice-cold buffer and mixed with percoll medium in an ultracentrifuge tube. The pure mitochondrial fraction settled at the bottom of the tube was recovered. The fraction was then diluted in buffer and centrifuged again at 10,000g (11,300rpm). Then mitochondrial lysis buffer (C36014, Beyotime) was added to the precipitate to extract mitochondrial proteins.
Western Blotting
Mitochondrial proteins were collected and prepared for western blotting (WB) detection. The protein concentrations were quantified using the bicinchoninic acid (BCA) assay kit provided by Beyotime (P0012). Protein extracts and molecular weight standards were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels, which were then transferred onto poly vinylidene fluoride (PVDF) membranes sourced from Bio-Rad (USA). After transfer, the PVDF membrane was incubated with 5% bovine serum albumin (BSA) blocking solution on a shaker for 1 h at room temperature. Then, the membrane was incubated with primary antibodies [PACS2, ab222316, Abcam; TRPV1, ab305299, Abcam; microtubule-associated protein 1 light chain 3 beta (MAP1LC3B), ab192890, Abcam; β-actinin, ab8226, Abcam] at 4℃ overnight. After being washed with Tris-Buffered Saline and Tween (TBST) buffer, the PVDF membrane was incubated with HRP-conjugated secondary antibody (ab288151, Abcam) at the recommended dilution, incubated at room temperature for 1 h, and washed with TBST buffer. Protein bands were detected with a chemiluminescence (ECL) chemiluminescent kit (P0018S, Beyotime) in a dark room and quantified using the Image Lab software (Version 6.0.1, Bio-Rad, USA).
Co-Immunoprecipitation
Cardiomyocytes were harvested and lysed with mild RIPA buffer (89901, Invitrogen) directly on the plate for 30 min for Co-Immunoprecipitation (co-IP). In the meantime, 50μl of Dynabeads Protein G (1003D, Invitrogen) were incubated with 3μg of antibody at room temperature for 1 h. Subsequently, the protein lysate was mixed with the beads-antibody complex and incubated overnight at 4°C. The beads were washed three times with lysis buffer. Bound proteins and 10% inputs were detected by WB.
Transmission Electron Microscopy Analysis
The myocardium was fixed in 2.5% glutaraldehyde and 2% paraformaldehyde at a temperature of 4℃. Subsequently, they were post-fixed in 1% osmium tetroxide for an additional 2 h. Following rinsing with PBS, the tissue samples were dehydrated through a graded series of ethanol concentrations and embedded in Epon. Thin sections, measuring 0.1um in thickness, were then stained with uranyl acetate and lead citrate. These sections were examined using a transmission electron microscope (Model H-7650, Hitachi, Japan).
Immunofluorescence Staining
To detect mitophagy, LV samples were combined and incubated with primary anti-MAP1LC3B antibody (OSM00022G, Thermofisher) and anti-translocase of mitochondrial outer membrane 20 (TOM20) antibody (ab186735, Abcam) at 4℃ overnight and then washed in PBS twice, before staining with the secondary antibodies (A11078, Thermo Fisher; ab150083, Abcam) at 37℃ for 2 h. The nucleus was stained with 4’,6-diamidino-2-phenylindole (DAPI) (C1002, Beyotime). Target protein expression was visualized under the confocal laser scanning microscope (LSCM) (A1R HD25, Nikon, Tokyo, Japan). Samples without primary antibodies were used as negative controls. Images were analyzed using ImageJ software (V1.8.0.112).
Oxygen Consumption Rate Measurement
The mitochondrial oxygen consumption rate (OCR) was evaluated using the Seahorse XF96 Analyzer (Seahorse Bioscience, North Billerica, USA). Briefly, cells were seeded in plates at a density of 8000 cells per well and incubated in XF assay medium without CO2 at 37℃ for 1 h. Subsequently, the cells were exposed to a cocktail of compounds, including 1µM oligomycin, 2µM trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP), and 1µM rotenone combined with antimycin, all provided in the XF Cell Mito Stress Test Kit (Seahorse Bioscience). Three measurements were taken during each cycle, and data on basal respiration, maximal respiration, and proton respiration were collected using the Seahorse XF96 Analyzer software, following the manufacturer’s recommended protocol. The spare respiratory capacity and adenosine triphosphate (ATP) production was calculated.
Measurement of Mitochondrial Calcium
Cardiomyocytes were seeded on glass-bottomed cell culture dishes and incubated with 1μM of the calcium indicator Rhod2-AM (ab142780, Abcam) at 37 °C in the dark for 30 min. Next, cells were washed twice with Hanks’ Balanced Salt Solution (HBSS) and imaged under a LSCM. The fluorescence intensity (F) was normalized to the baseline fluorescence value F0 (F/F0) and expressed as mitochondrial calcium concentration ([Ca2+]m). We measured Fmax and Fmin, as previously described. Fmax was obtained by perfusion with 10μM ionomycin and 5mM CaCl2; Fmin was measured by perfusion with 10mM EGTA and 2μM BAPTA-AM (B1205, Molecular probes) in HBSS. Capsazepine (211,280, Sigma-Aldrich) and thapsigargin (TG) (T9033, Sigma-Aldrich) were also added to the external solution at a proper final concentration. The fluorescence intensity was converted to [Ca2+] using the following formula: [Ca2+]m = Kd × (F − Fmin)/(Fmax − F), where Kd is the equilibrium dissociation constant of Rhod2 for Ca2+, which was 570nM.
Real-Time Quantitative PCR
To quantify miRNA, total RNA was extracted from tissue samples using the RNAiso Small RNA Kit (9753A, TAKARA, Japan), strictly adhering to the guidelines. The extraction process involved obtaining total RNA from 50-100mg of myocardial tissue, to which RNAiso was subsequently added. The tissue was then homogenized. For the synthesis of cDNA, 1ug of the extracted total RNA was utilized with the Mir-X™ miRNA First-Strand Synthesis Kit (638,313, TAKARA, Japan). The qRT-PCR reactions were conducted using the SYBR Premix Ex Taq™ II Kit (A46109, Thermofisher, USA). All reactions were performed in triplicate. The relative expression levels of target miRNAs were normalized to U6 snRNA using the 2^(-ΔΔCt) method.
Dual-Luciferase Assay
Isolated cardiomyocytes were cultured and transfected with plasmids expressing wild-type or mutant 3’UTR fragment luciferase reporters (500ng per well). Forty-eight hours post-transfection, luciferase activity was measured using a GloMax-Multi Detection System (E1960, Lumiprobe, US) via the Promega Dual-Luciferase system (E1910, Promega Corporation, USA) according to the manufacturer's instructions [18].
Tail Vein Administration of AntagomiR-499-5p
Fifty nmol of Antagomir-499-5p [Anti-miR-499-5p (5’-AAACAUCACUGCAAGUCUUAA-3’), RIBOBIO, China] was dissolved in 375uL of ddH2O, as per the manufacturer’s guidelines. Subsequently, 375uL of a 10% glucose solution was introduced and thoroughly mixed to prepare Reagent A. Reagent B was then created by combining 375uL of Entranster TM-in vivo transfection reagent with an equal volume of 10% glucose solution. The working solution was ultimately obtained by merging Reagents A and B in a 1:1 ratio. Mice were administered 300uL of the working solution (~ 10nmol of antimiR-499-5p or anti-miR-NC) via tail vein injection.
Generation of Cardiomyocyte-Specific Pacs2 Knock-in Mice
The cardiomyocyte-specific Pacs2 knock-in in C57BL/6N mice were generated using the CRISPR/Cas9 system. The Hipp11 (H11) locus is located within an intergenic region between the Eif4enif1 and Drg1 genes on mouse chromosome 11 (~ 0.7kb 5’ of Eif4enif1 gene and ~ 4.5kb 3’ of the Drg1 gene). The “ɑ-MHC_long promoter-Kozak-Mouse Pacs2 CDS-rBG pA” cassette will be inserted into H11 locus. To engineer the targeting vector, homology arms will be generated by PCR using BAC clone as template. Cas9 and gRNA will be co-injected into fertilized eggs with targeting vector for mice production.
MiRNA Sequencing and Data Analysis
Total RNA was extracted from myocardial tissue, and the small RNAs within were detected by Nuohe Biological Co., Ltd. The construction and sequencing of the small RNA library were also performed by Nuohe Biological Co., Ltd. Subsequently, the cDNA library underwent sequencing on HiSeq 2500 System, Illumina, Inc. Raw data were compiled using Illumina analysis system. In the data analysis phase, the threshold for significantly differential expression was established at a p-value of less than 0.05 and an absolute log2(fold change) greater than 1. R packages (Version 4.3.3) were utilized to generate a volcano plot which was used to display the differentially expressed miRNAs.
ITRAQ Proteomics Analysis
The protein from the samples was extracted and precipitated by adding acetone and leaving it overnight at −20 ℃, followed by washing. Sequence grade modified trypsin (Promega, Madison, WI) was added to digest the proteins at 37℃ overnight. The peptide mixture was desalted by C18 ZipTip, quantified by Pierce™ Quantitative Colorimetric Peptide Assay, and lyophilized by SpeedVac. The resultant peptide mixture was labeled with iTRAQ 8Plex labeling kit (Sciex) and then were pooled in a vacuum concentrator. The peptide mixture was re-dissolved and then fractionated by high pH separation connected to a reverse phase (XBridge C18) column (Waters Corporation, MA, USA). Then the peptides were analyzed by on-line nanospray LC–MS/MS on Q Exactive™ HF-X coupled to EASY-nLC 1200 system (Thermo Fisher Scientific, MA, USA). Data analyse were processed by Shanghai Genechem Co., Ltd. Differentially expressed proteins were filtered if they contained ≥ 1 unique peptide with p-value less than 0.05 and fold change ≥ 2.
Protein Docking
The target protein structures were retrieved from the UniProtKB database and subsequently input into the GRAMM system. The computational process was initiated using the platform's default protein–protein docking parameters. Upon completion of the program, the theoretical optimal binding conformation was collected from the system's output. Binding energy was used as the primary evaluation metric, and an in-depth analysis of the optimal docking conformation was conducted, taking into account characteristics such as the protein interaction region surface area, hydrogen bonding interactions, and key amino acid residues. After the docking process was completed, PDBePISA was utilized to calculate the binding free energy, and gPyMOL (version 3.1) was employed for visualization analysis.
Statistical Analysis
Continuous data were presented as the mean ± standard deviation, unless otherwise stated. Statistical comparisons between two or more groups were conducted using the unpaired two-tailed Student’s t-test or for two-group comparisons, one-way ANOVA with Tukey’s post-hoc test for multiple groups. All statistical analyses were performed using the software SPSS 27.0 and Prism 10.1.12. Statistical significance was set at *P < 0.05, **P < 0.01 and ***P < 0.001.

Comments (0)