
Remember me

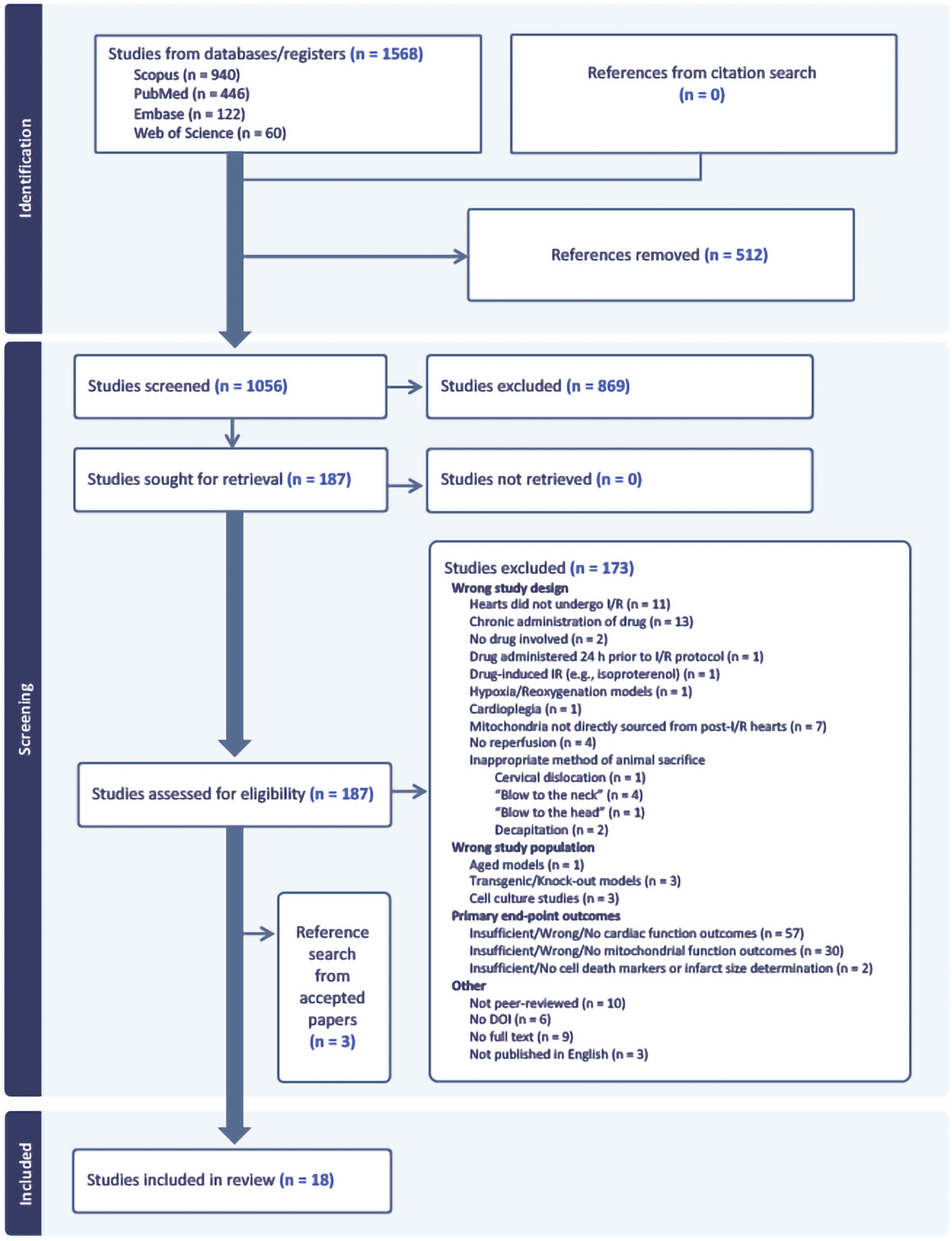
Eighteen papers were included in the review from 1571 studies (1568 papers, including three studies from citation search in accepted papers), as detailed in the PRISMA flow diagram (Fig. 1). Interestingly, all included studies investigated male rodents only, with 94% of studies completed using rats, and a single study using C57BL/6J mice. Wistar rats were the most studied rat strain (53%), followed by Sprague-Dawley rats (41%), and one study was on Fischer-344 rats. Body weight was reported across 94% of the studies and ranged from 140 to 420 g for rats and 25 to 35 g for mice. Only 28% of studies reported age which ranged between 4 and 34 weeks old. Despite not filtering animal species, only rodent studies fit the inclusion criteria. Studies including other species and larger animals were identified but were rejected due to having insufficient mitochondrial and cardiac data. Animal characteristics are summarised in SI 3.
Fig. 1
PRISMA flow diagram. Studies from Scopus, PubMed, Embase, and Web of Science were screened according to exclusion and inclusion criteria
Drug Characteristics and Ischaemia-Reperfusion ModelsThe primary drugs of interest and their optimal doses were extracted from each study (SI 4). These included sodium hydrogen sulfide (NaHS, 20 µM), 5-azacytidine (5 µM), sodium thiosulfate (STS, 1 mM, n = 4 studies), ARP-100 (10 µM), ONO-4817 (50 µM), amobarbital (2.5 mM), AVE-4890 (5 µM), angiotensin II (10 nM), metformin (200 mg/kg), diazoxide (100 µM), sappanone A (100 µM), cariporide (10 µM), metoprolol (10 µM), 3,4-dihydroxy-phenyl lactic acid (DLA, 5 mg/kg), and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO, 1 mM). Sixteen studies used the Langendorff isolated heart model to assess the cardioprotective efficacy of the compounds via intracoronary perfusion. The remaining two studies utilised an in vivo model of I/R induced by left anterior descending coronary artery ligation with intravenous drug delivery. The ischaemic period ranged from 25 to 40 min, and the reperfusion period ranged from 30 to 120 min. Drug administration was performed either before prolonged ischaemia, or during ischaemia, or both.
Primary outcome measures were cardiac functional recovery and cellular injury (Table 2), and mitochondrial oxygen consumption, enzymatic activity and complex expression (Table 3). The summary of outcomes from the included studies focuses on comparisons of the individual effects of the primary drugs of interest alone versus the control I/R group. Co-treatment with another drug, or a more established I/R therapy (e.g., ischaemic preconditioning or ischaemic postconditioning), is occasionally mentioned when relevant to the interpretation of results. The most frequently recorded cardiac functional response was left ventricular developed pressure (LVDP) (89%, including those reported LVDP as a percentage of baseline function), followed by rate pressure product (61%, RPP), left ventricular end-diastolic pressure (56%, LVEDP), and rates of contraction and relaxation (50%, maximum and minimum dP/dt). Other parameters investigated from the ex vivo models were left ventricular end-systolic pressure (LVESP) and ischaemic contracture. Alongside these parameters, in vivo models also assessed arrhythmia scores, mean arterial pressure, stroke volume and ejection fraction after I/R. The most recorded mitochondrial outcome within the inclusion criteria was mitochondrial ETC activity (61% of studies), followed by mitochondrial respiration (56% of studies) and mitochondrial ETC protein expression (28% of studies). Other mitochondrial parameters, such as mitochondrial swelling, mitochondrial membrane potential, and other mitochondrial-related pathways and upstream pathways, were noted but were not considered primary outcomes. To aid interpretation of primary outcomes, results were synthesised based on shared pharmacological effects across cardiac, cellular, and mitochondrial outcomes rather than organised by individual studies. To support this, an integrated summary of mechanistic outcomes across all compounds is provided in Table 4. In terms of their classification according to their effect on mitochondrial function, compounds were categorised as either:
Table 2 Cardiac and cell death outcome following I/RTable 3 Mitochondrial outcomes following I/RDirect regulators of mitochondrial bioenergetics (e.g. reversible complex I or complex II inhibition).
Indirect regulators of mitochondrial function, which target mitochondrial dynamics, redox signalling, exhibit antioxidant and free radical scavenging activities, and preserve mitochondrial function by targeting upstream canonical pathways of cardioprotection.
Effects of Pharmacological Interventions on Cardiac Function and Cell Death ParametersDirect Modulators of Mitochondrial BioenergeticsAmobarbital and diazoxide enhanced cardiac outcomes following ischaemia in Fischer-344 and Sprague-Dawley rats, respectively, when administered before ischaemia. Amobarbital, a reversible complex I inhibitor enhanced contractile function by a significant increase in LVDP, RPP and the heart’s rate of relaxation and contraction at the end of reperfusion when perfused 1 min before prolonged ischaemia [27]. This was accompanied by decreased diastolic pressure, infarct size and LDH levels at the end of reperfusion. Diazoxide (100 µM) reversibly inhibited complex II, delayed ischaemic contracture, and increased LVDP and RPP. This was associated with a decrease in LVEDP and a 73% reduction in infarct size at the end of reperfusion [28].
Indirect Modulators of Mitochondrial Function Sulfide DonorsSulfide-based drugs NaHS (20 μm) and STS (1 mM) consistently increased recovery of cardiac parameters while predominantly decreasing cell death markers in male Wistar rats. Both NaHS and STS administered before and after ischaemia resulted in significantly increased developed pressure, RPP, and in studies that measured it, systolic pressure, alongside reduced LVEDP at the end of reperfusion [29,30,31,32,33,34]. This was accompanied by a significant reduction in infarct size and lowered cell injury markers including lactate dehydrogenase (LDH) and creatine kinase (CK), while in studies that assessed DNA fragmentation, no damage was detected [29, 30, 34]. The timing of STS administration influenced cell death outcomes. For example, LDH levels were unaffected by STS postconditioning [34] but were reduced by STS preconditioning [31]. Overall, sulfide donors increased recovery of the heart following I/R and predominantly decreased necrotic cell death.
Ion Handling ModulatorsPre-ischaemic and post-ischaemic administration of AVE-4890, an NHE-1 inhibitor, was reported to also increase LVDP and decrease LVEDP at the end of reperfusion in isolated Sprague-Dawley rats hearts subjected to I/R [35]. It caused a significant reduction in LDH levels and in apoptotic markers such as cleaved poly-ADP ribose polymerase (PARP), but levels of caspase-3 remained unchanged compared to the I/R group in the cytosolic fraction. Despite this, other markers of apoptosis, such as apoptotic inducing factor and EndoG, remained in the mitochondria and had elevated protein expression levels in the organelle. Cariporide (10 µM) and metoprolol (10 µM), a sodium-hydrogen antiporter 1 (NHE-1) inhibitor and a beta-blocker, were used individually and in combination as a treatment against I/R injury in isolated Sprague-Dawley rat hearts [36]. Wang et al. found that I/R hearts exposed to either treatment had similar increases in LVDP and the rate of myocardial contraction and relaxation with a consequent decrease in diastolic pressure. Investigators also reported a reduction in infarct size for cariporide or metoprolol alone, but the effect was significant in I/R groups exposed to metoprolol only, but not cariporide alone. Collectively, treatments that prevented ion imbalance reduced cardiac dysfunction and prevented irreversible tissue damage.
Antioxidant/Free Radical ScavengersTreatment with DLA at a 5 mg/kg dose in an in vivo I/R study using Sprague-Dawley rats resulted in attenuated infarct size and apoptosis markers [37]. However, the levels of coronary LDH release remained unchanged and comparable to those of the I/R group. Nevertheless, anti-apoptotic protein expression of B-cell lymphoma 2 (Bcl-2) was increased while pro-apoptotic Bax protein levels were diminished. This corresponded with the restoration of the Bcl-2/Bax ratio by DLA treatment. Additionally, DLA increased left-ventricular maximum and minimum developed pressures, leading to a reduction in end-diastolic pressure. Other cardiac functional parameters, such as mean arterial pressure and systolic pressure, were also restored by DLA treatment. In a study in which the free-radical scavenger spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO), was administered at a concentration of 1 mM before ischaemia and during reperfusion to test its cardioprotective potential, it increased LVDP and RPP and significantly lowered LVEDP and infarct size in isolated Sprague-Dawley rat hearts [38].
Matrix Metalloproteinase InhibitorsMatrix metalloproteinase-2 (MMPs) are endopeptidases activated by oxidative stress during I/R through proteolytic cleavage, resulting in NTT-MMP-2, a truncated isoform of MMP. Bassiouni et al. hypothesised that the NTT-MMP-2 isoform proteolyzes Mfn-2 during I/R [39]. Inhibitors specific to MMP, such as ARP-100 and ONO-4817, were used to test this hypothesis in male C57Bl/6J mice and showed significant improvements in LVDP and rates of left ventricular contraction and relaxation. A concomitant attenuation of LDH levels was also seen at the end of reperfusion.
Canonical Pathway and Metabolic Status ModulatorsMetformin preserved cardiac functional recovery and reduced cell death in ex vivo and in vivo models of I/R. In isolated Sprague-Dawley rat hearts, metformin (2 mM) was perfused before ischaemia and throughout reperfusion, resulting in an overall increase in LVDP and RPP at the end of reperfusion [40]. In contrast, Palee et al. administered metformin (200 mg/kg) intravenously via the femoral vein in Wistar rats 15 min before coronary artery ligation. Echocardiography demonstrated significant improvements in RPP, LVESP, ejection fraction, and stroke volume, accompanied by reductions in LVEDP and arrhythmia score [41]. However, metformin did not affect the rates of contraction and relaxation. Despite differences in experimental model, route, and timing of administration, metformin lowered LDH levels, infarct size, and apoptotic markers, including caspase-3 and TUNEL-positive cells. Other apoptotic markers, such as Bcl-2 protein expression, remained unchanged, whereas Bax was significantly increased in the metformin groups compared with the control I/R group.
Similarly, pre-ischaemic perfusion with 5-azacytidine (5 µM) in isolated Wistar rat hearts increased LVDP and RPP, including the rate of contraction and relaxation of the I/R heart at the end of reperfusion. It also reduced infarct size, significantly attenuated LDH and CK levels, and lowered caspase-3 activity. Angiotensin II also increased post-ischaemic LVDP, RPP and rate of contraction, while ischaemic contracture was unaffected. Additionally, angiotensin II decreased both infarct size and LDH significantly [38]. On the other hand, sappanone A (100 µM) post-conditioning increased LVDP and the heart’s rates of contraction and relaxation, and significantly reduced cell death markers such as LDH, CK, and cardiac troponin I (cTnI) at the end of reperfusion [42].
Effects of Pharmacological Interventions on Mitochondrial BioenergeticsMitochondrial Oxygen ConsumptionSeveral compounds enhanced complex I-driven mitochondrial oxygen consumption, including NaHS, amobarbital metformin, 5-azacytidine, ARP-100, ONO-4817, and metoprolol. This was shown in increased State 3 respiration, respiratory control ratios (RCR) and adenosine diphosphate/oxygen (ADP/O) ratios [27, 29, 30, 36, 39,40,41, 43]. In studies using NaHS, complex I function was restored to near baseline levels, but did not yield a statistical difference when compared with I/R. This effect was more prominent in IFM than SSM. In contrast, treatment with 5-azacytidine significantly augmented complex I-driven respiration in both the IFM and SSM. Amobarbital treatment transiently inhibited complex I during ischaemia and preserved mitochondrial oxidative phosphorylation following I/R, evidenced by significantly higher complex I–linked State 3 respiration and RCR compared with the I/R group in both SSM and IFM [27]. These values were significantly higher than the I/R group but remained below the non-ischaemic time control group. Metalloproteinase inhibitors ARP-100 and ONO-4817 also increased complex-I driven state 3 respiration and RCR. In contrast, angiotensin II caused a non-significant increase in complex-I-driven respiration, with no impact on the RCI compared with the I/R group when administered alone [44].
Fewer compounds investigated respiration via complex II and complex IV. Complex II-driven RCR in both mitochondrial subpopulations remained unchanged with 5-azacytidine, with only the ADP/O ratio of the IFM responding favourably to the drug [43]. In contrast, AVE-4890, ARP-100, and ONO-4817 significantly increased complex II-driven State 3 respiration and associated RCR and RCI. Metformin also enhanced complex IV respiration accompanied by increases RCI and ADP/O ratios [40, 41]. Among the compounds, only cariporide did not affect mitochondrial oxygen consumption. Though, it increases complex I-linked respiration when combined with metoprolol.
Mitochondrial ETC Complex ActivityAcross the included studies, several compounds improved activities of ETC complexes I–IV. Complex I activity was the most consistently enhanced target. NaHS restored complex I activity in both mitochondrial populations to near baseline levels, with respirasome activities (I + III and II + III) restored primarily in the IFM [29, 30]. STS likewise increased complex I activity, although the mitochondrial compartment affected differed by timing: postconditioning increased complex I activity [34], whereas pre-ischaemic STS improved complex I activity predominantly in the SSM [31]. Additional studies using STS reported broader improvements in enzyme activities across complexes I–IV under both pre- and postconditioning conditions [32, 33].
Other compounds also demonstrated improvements in complex I-linked activity. Treatment with 5-Azacytidine increased the activities of complex I and IV in both IFM and SSM [43]. On the other hand, amobarbital transiently inhibited complex I during ischaemia and preserved complex I activity and corresponding I + III respirasome activity in both mitochondrial subpopulations [27]. DLA restored complex I activity by enhancing NDUFA10 expression [37], while DMPO also increased the activities of complexes I and IV [38], and sappanone A increased complex I–IV activities [42]. Complex II activity was less consistently modulated. Treatment with NaHS did not restore complex II activity in either mitochondrial subpopulation [26] while STS decreased complex II activity despite improving complexes I, III, and IV [31, 34]. Diazoxide reversibly inhibited complex II during ischaemia, with activity returning upon reperfusion [28].
Mitochondrial ETC Protein ExpressionFewer studies assessed protein expression of ETC complexes. Administration of STS produced mixed effects: preconditioning decreased complex I and II protein levels, while complex V expression remained unchanged [31]. DLA increased complex I subunit NDUFA10 expression alongside enhanced complex I activity [37]. DMPO increased protein expression of complexes I, II, and IV [38] while metformin only enhanced complex V protein expression but not complex I to complex IV.
Influence of Upstream Pathways and Other Mitochondrial ParametersMost studies modulated mitochondrial function indirectly through upstream cardioprotective signalling. The cardioprotective effects of STS preconditioning were attenuated when phosphoinositide 3-kinase (PI3K), mammalian target of rapamycin (mTOR), and mitochondrial ATP-sensitive potassium (mito-KATP) channels were inhibited, suggesting a dependence on these pathways [33]. The molecular mechanisms underlying STS-mediated cardioprotection may differ between preconditioning or postconditioning protocols and require further investigation [33]. Similarly, 5-azacytidine involves PI3K, glycogen synthase kinase 3β (GSK3β), and the mito-KATP pathway [43], while the cardioprotective effects of DLA were abolished by sirtuin 1 (SIRT1) inhibition using sirtinol or EX-527 [37]. Metformin’s cardioprotective effect also differed between ex vivo and in vivo models. The effects of metformin in the ex vivo model were abolished by PPARα inhibition [40], while the in vivo model was dependent on AMPK, connexin-43 phosphorylation, PGC-1α upregulation and reduced Drp1 translocation [41]. On the other hand, angiotensin II improved cardiac recovery when administered alone by binding to AT-1 receptors but did not improve mitochondrial function [44]. However, when paired with IPC, angiotensin II improved complex I respiration by activating PKC.
Several studies also assessed additional mitochondrial parameters relevant to I/R injury, including outer membrane integrity, mitochondrial dynamics, mitochondrial biogenesis, mitochondrial calcium content and resistance to mPTP opening during I/R. Sappanone A, prevented mPTP opening, reduced mitochondrial ROS, promoted a shift toward mitochondrial fusion and modulated mitophagy markers, including reductions in PINK1 and p62 and increases in Parkin and LC3II in an AMPK-dependent [42]. Administration of STS before ischaemia increased mitochondrial copy number and mRNA levels of genes involved in ATP synthesis, consistent with increased PGC1α expression. On the other hand, MMP inhibitors increased ATP production during respiration (measured via fluorometric assay of respiration buffer after respirometry) and preserved mitochondrial mitofusin-2 (Mfn-2) protein levels [39]. Amobarbital preserved cytochrome c content, a marker of outer membrane integrity, in both the SSM and IFM [27]. In contrast, the cardioprotective effects of diazoxide were abolished by mito-KATP channel inhibitor 5-hydroxydecanoic acid (5-HD) [28]. Neither β-blockade with metoprolol nor NHE-1 inhibition with cariporide significantly reduced mitochondrial calcium accumulation following I/R. Overall, pharmacological agents target diverse upstream and other mitochondrial-related pathways, such as PI3K/Akt, mTOR, AMPK, mito-KATP channels, PGC1α, PPARα, SIRT1, and AT1R signalling, calcium handling and the mPTP (Table 4).
Table 4 Mechanistic classification of pharmacological drugs targeting mitochondrial function during I/R
Comments (0)