Cell culture and grouping treatment
Human rheumatoid arthritis fibroblast-like synoviocytes (MH7A cells) (BNCC371792, BeNa Culture Collection) were cultured in complete Dulbecco’s Modified Eagle Medium (DMEM) (Kaiji Biology, KGL1212-500, containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin) in a 37 °C incubator with 5% CO₂ (BPN-80CW, Shanghai Yiheng Scientific Instrument Co., Ltd.). All experiments used cells at passages 5–8 in the logarithmic growth phase to avoid phenotype drift.
Cells were subjected to reagent intervention in groups according to the requirements of subsequent experiments. Based on the results of preliminary experiments, the concentration of FBS in DMEM medium was reduced to 2% during drug intervention in all groups to minimize the interference of cell proliferation effects on the experimental results.
Control group
MH7A cells treated with low-serum DMEM medium containing eATP (0 mM) for 24 h.
eATP group
MH7A cells treated with low-serum DMEM medium containing eATP (2 mM) for 24 h.
eATP + A804598 group
MH7A cells pre-incubated in low-serum DMEM medium containing A804598 (50 µM) for 2 h first, then treated with fresh low-serum DMEM medium containing both A804598 (50 µM) and eATP (2 mM) for 24 h.
A804598 group
MH7A cells pre-incubated in low-serum DMEM medium containing A804598 (50 μM) for 2 h first, then treated with fresh low-serum DMEM medium containing A804598 (50 µM) only for 24 h.
Cell viability detection by CCK-8 assay
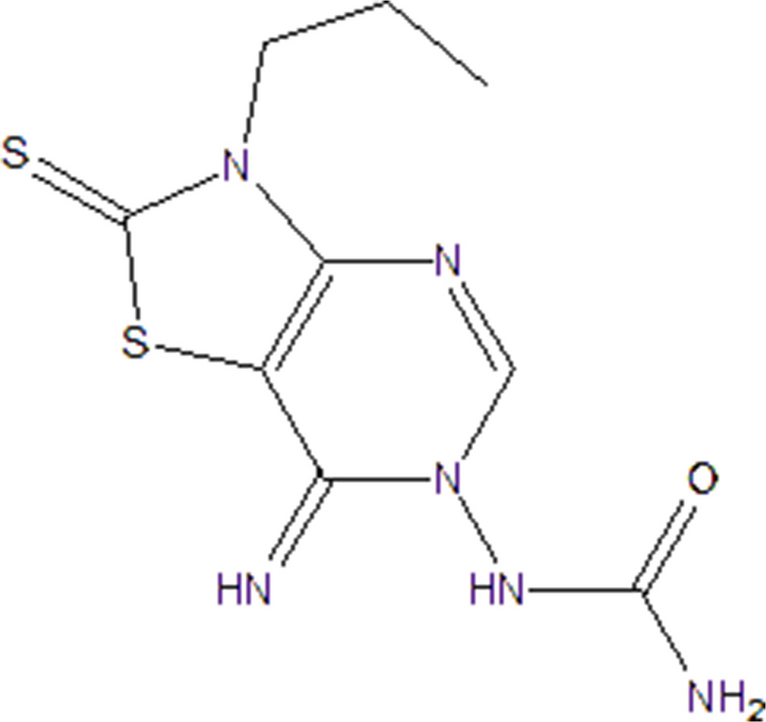
Passaged MH7A cells were digested with trypsin and seeded into 96-well plates. The cells were treated with complete DMEM medium containing eATP at different concentrations (0, 0.1, 0.2, 0.4, 0.8, 1, 2, 4, 8 mM) for 24 h. Another group of cells was treated with complete DMEM medium containing P2X7R competitive antagonist (A804598, MCE) at different concentrations (0, 2.5, 5, 10, 20, 50, 100 µM) for 24 h. Subsequently, 10 µL of CCK-8 reagent was added to each well, and the cells were incubated at 37 °C for 2 h. The absorbance at 450 nm was measured using a microplate reader (Prolong, China) to calculate cell metabolic viability, and the appropriate concentration was selected for subsequent experiments.
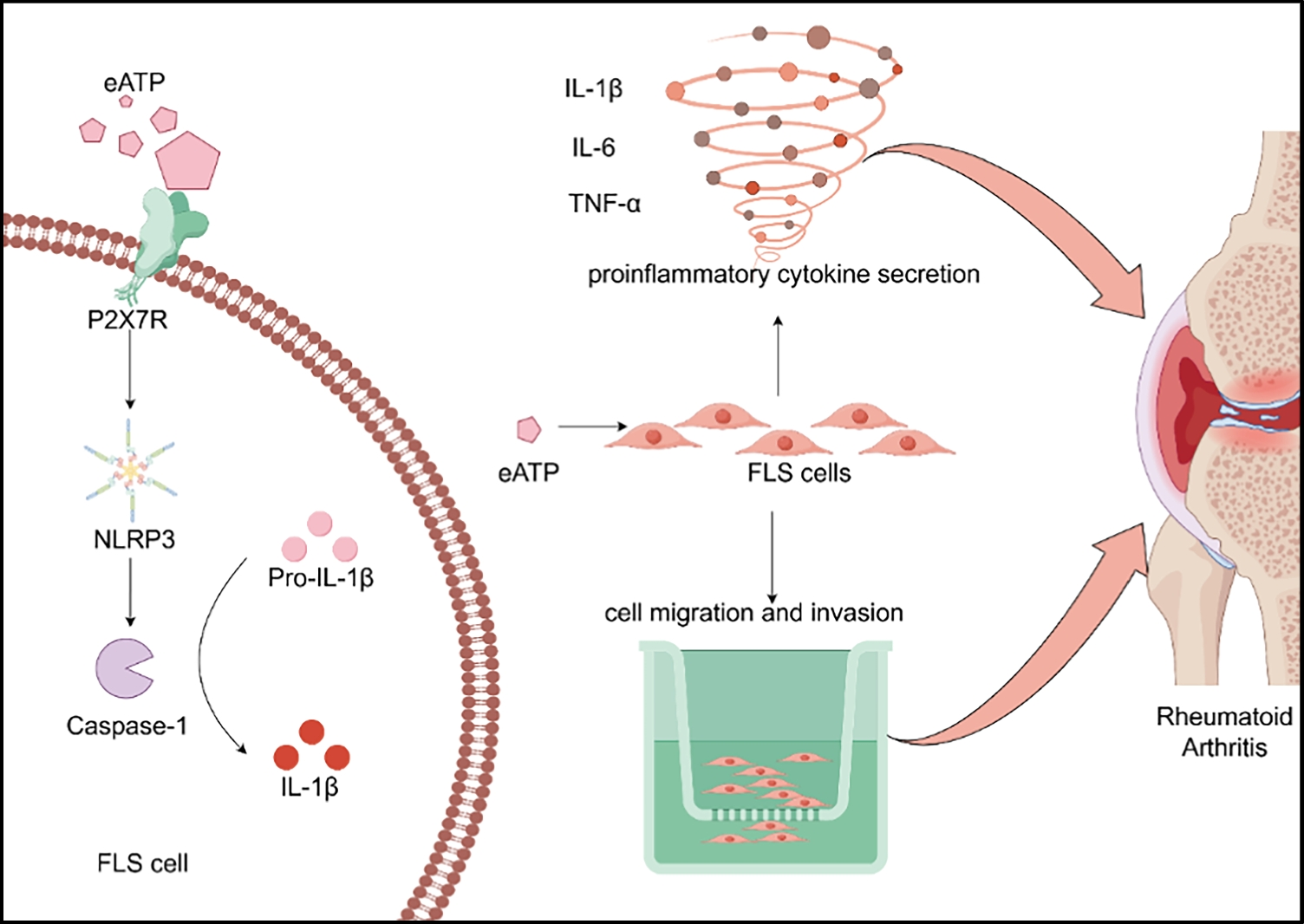
Cell migration and invasion assaysMigration assay
Cells were seeded into 24-well plates as required. After digestion and dilution, 0.3 mL of cell suspension (cell density: 8 × 104 cells/well) in serum-free basic medium was added to the upper chamber of Transwell inserts, and 0.5 mL of complete medium was added to the lower chamber. The plates were incubated in the incubator for 24 h and 48 h. After incubation, the inserts were taken out, the medium was discarded, and the cells were stained with 0.1% crystal violet (G1061, Solarbio) overnight. Cells on the inner surface of the upper chamber were wiped off with cotton swabs, and the inserts were observed under a microscope (BX43, Olympus). After photographing, the staining solution was removed, 800 µL of 33% acetic acid was added, and the mixture was fully mixed and allowed to stand. Then, 200 µL of the solution from each well was transferred to a 96-well plate, and the absorbance was measured at 562 nm using a microplate reader (WD-2012B, Beijing Liuyi Instrument Factory).
Invasion assay
Cells were seeded into 24-well invasion plates as required. After digestion and dilution, 50 µL of Matrigel was first added to the upper chamber of the invasion inserts and allowed to solidify. Then, 0.3 mL of cell suspension (cell density: 8 × 104 cells/well) in serum-free basic medium was added to the upper chamber, and 0.5 mL of complete medium was added to the lower chamber. The plates were incubated in the incubator for 48 h. After incubation, the inserts were taken out, the medium was discarded, and the cells were stained with 0.1% crystal violet (G1061, Solarbio) overnight. Cells on the inner surface of the upper chamber were wiped off with cotton swabs, and the inserts were observed under a microscope (BX43, Olympus). After photographing, the staining solution was removed, 800 µL of 33% acetic acid was added, and the mixture was fully mixed and allowed to stand. Then, 200 µL of the solution from each well was transferred to a 96-well plate, and the absorbance was measured at 562 nm using a microplate reader (WD-2012B, Beijing Liuyi Instrument Factory).
qPCR detection
Total RNA was extracted from cells using Trizon reagent (CW0580S, CWBIO). mRNA and miRNA were extracted using an RNA ultra-pure extraction kit (CW0581M, CWBIO) and a miRNA ultra-pure extraction kit (CW0627S, CWBIO), respectively. The concentration and purity (OD260/OD280) of mRNA and miRNA were determined using a UV-visible spectrophotometer (NP80, NanoPhotometer). cDNA was synthesized using an RNA reverse transcription kit (R223-01, Vazyme) and a miRNA reverse transcription kit (MR101-02, Vazyme). Quantitative real-time PCR was performed using a fluorescent PCR instrument (CFX Connect™ Real-Time System, Bio-Rad Laboratories (Shanghai) Co., Ltd.). The reaction procedure was as follows: pre-denaturation at 95 °C for 10 min; denaturation at 95 °C for 10 s, annealing at 58 °C for 30 s, and extension at 72 °C for 30 s, with 40 cycles. β-actin and U6 were used as internal references, and the relative expression level of genes was calculated using the 2-△△Ct method. The primer sequences are shown in Table 1.
ELISA detection
The expression levels of TNF-α, IL-1β, and IL-6 in the cell culture supernatant of each group were detected using a human tumor necrosis factor-α (TNF-α) kit (E-EL-H0109, Elabscience), a human interleukin-1β (IL-1β) kit (E-EL-H0149, Elabscience), and a human interleukin-6 (IL-6) kit (E-EL-H6156, Elabscience), respectively. The cell supernatant was centrifuged at 1000×g for 20 min to remove impurities and cell debris, and the supernatant was collected for detection. The specific operation steps were as follows: the ELISA kit was equilibrated at room temperature, and the standard solution was prepared according to the instructions. Blank wells, standard wells, and sample wells were set up. No sample or enzyme-labeled reagent was added to the blank wells, while 50 µL of sample or standard solution was added to the other wells, followed by 100 µL of enzyme-labeled reagent. The plates were incubated at 37 °C for 1 h. After washing the plates 5 times, the chromogenic reagent was added, and the plates were incubated at 37 °C for 15 min. The reaction was terminated by adding the stop solution, and the absorbance of each well was measured at 450 nm using a microplate reader (WD-2012B, Beijing Liuyi Instrument Factory).
Western blot
Cells were collected, and the medium was discarded. Total protein was extracted by lysis on ice using RIPA lysis buffer (C1053, Beijing Pulilai Gene Technology Co., Ltd.). The lysate was centrifuged at 12,000 r/min at 4 °C for 10 min using a high-speed centrifuge (5424R, Eppendorf), and the supernatant was collected. The total protein concentration was determined using a BCA protein quantification kit (E-BC-K318-M, Elabscience). After denaturation of the protein samples, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed for 1.5 h, followed by constant current transfer (300 mA) to a polyvinylidene fluoride (PVDF) membrane (Millipore) for 1–2 h. The PVDF membrane was blocked with skimmed milk, then incubated with primary antibodies at 4 °C overnight. On the next day, the PVDF membrane was incubated with secondary antibodies at room temperature for 2 h. The PVDF membrane was soaked in ultra-sensitive chemiluminescent solution (RJ239676, Thermo Fisher Scientific), and images were acquired using an ultra-sensitive chemiluminescence imaging system (Tanon-5200, Shanghai Tianneng Technology Co., Ltd.). The antibodies used and their corresponding dilution ratios are shown in Table 2.
Table 2 Antibody informationTo precisely distinguish between the precursor and activated forms of caspase-1, two specific antibodies were employed in the blotting analysis: (1) a rabbit monoclonal anti pro-caspase-1 + p10 + p12 antibody (ab179515, Abcam), which recognizes both the pro-caspase-1 precursor and its cleaved p10/p12 subunits; and (2) a rabbit polyclonal anti cleaved-caspase-1 antibody (AF4005, Affinity), which is specific to the activated, cleaved form of caspase-1. The use of these two antibodies allows for independent and specific verification of caspase-1 activation, thereby providing a more robust assessment of NLRP3 inflammasome activity.
Statistical analysis
GraphPad Prism 9.5 software was used for graphing and statistical analysis. Quantitative results were expressed as mean ± standard deviation (x ± s). Independent sample t-test was used for comparison between two groups; one-way analysis of variance (ANOVA) was used for comparison of quantitative data among multiple groups, followed by Tukey’s post hoc test. A P-value < 0.05 was considered statistically significant.


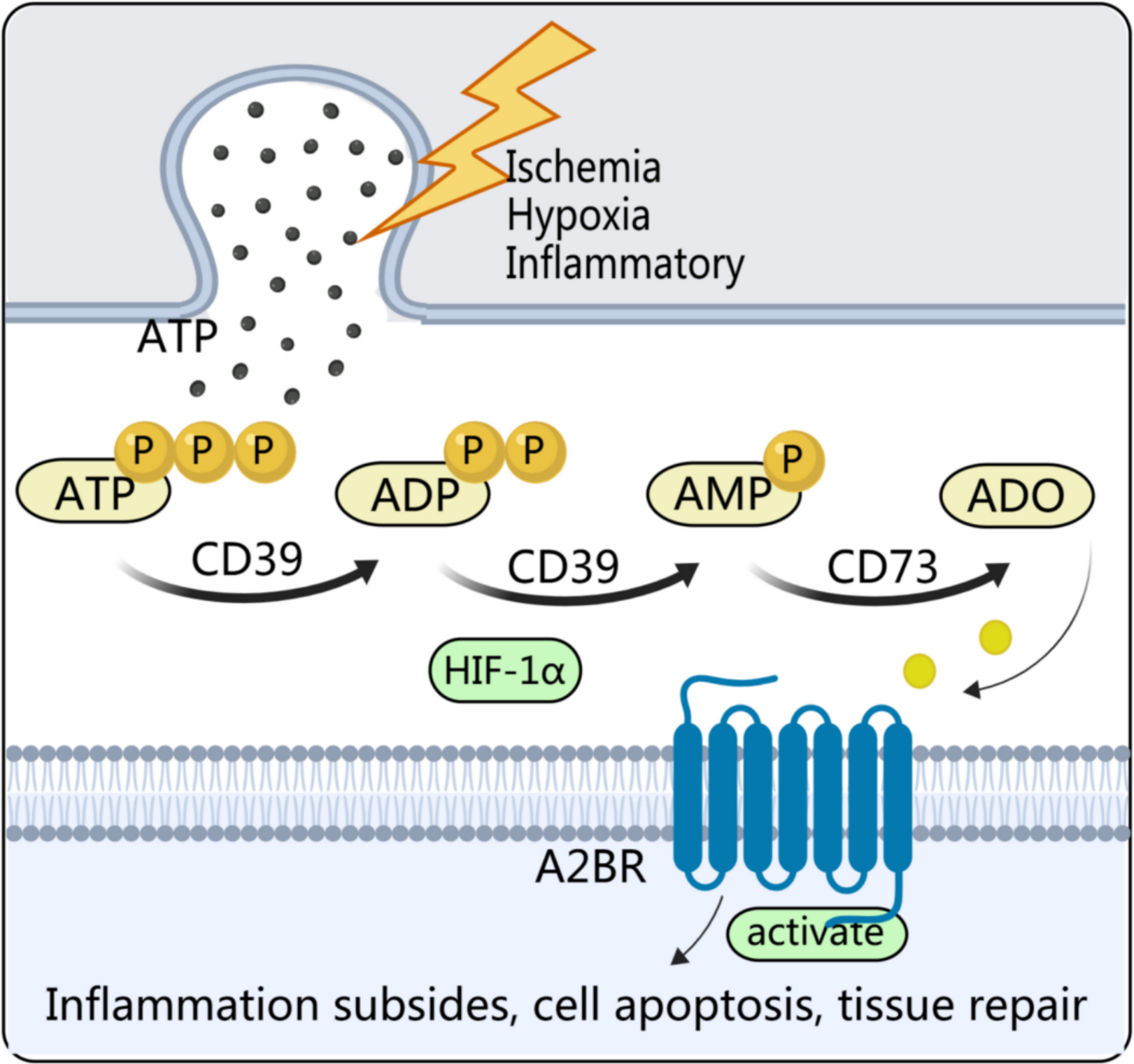
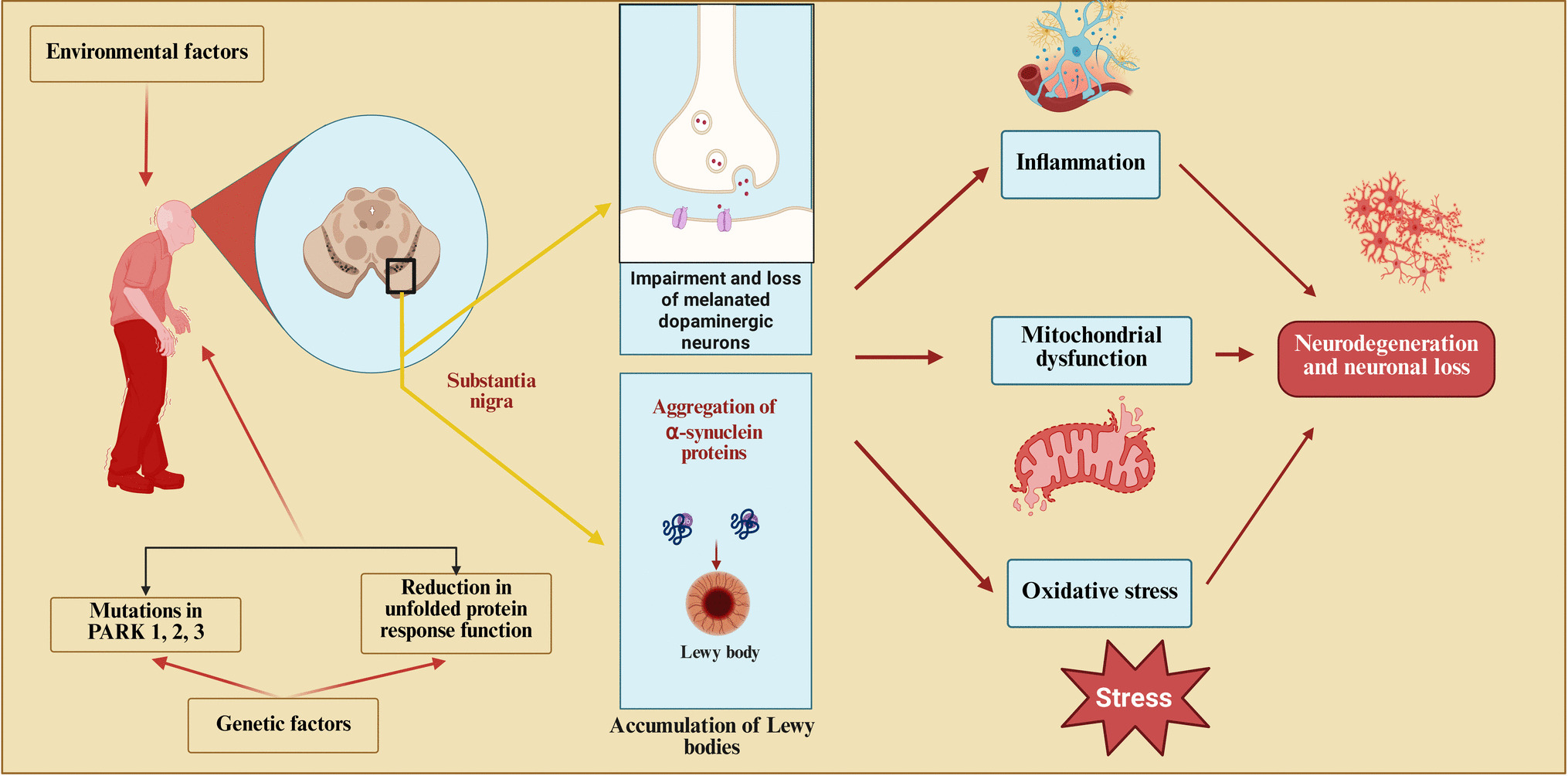
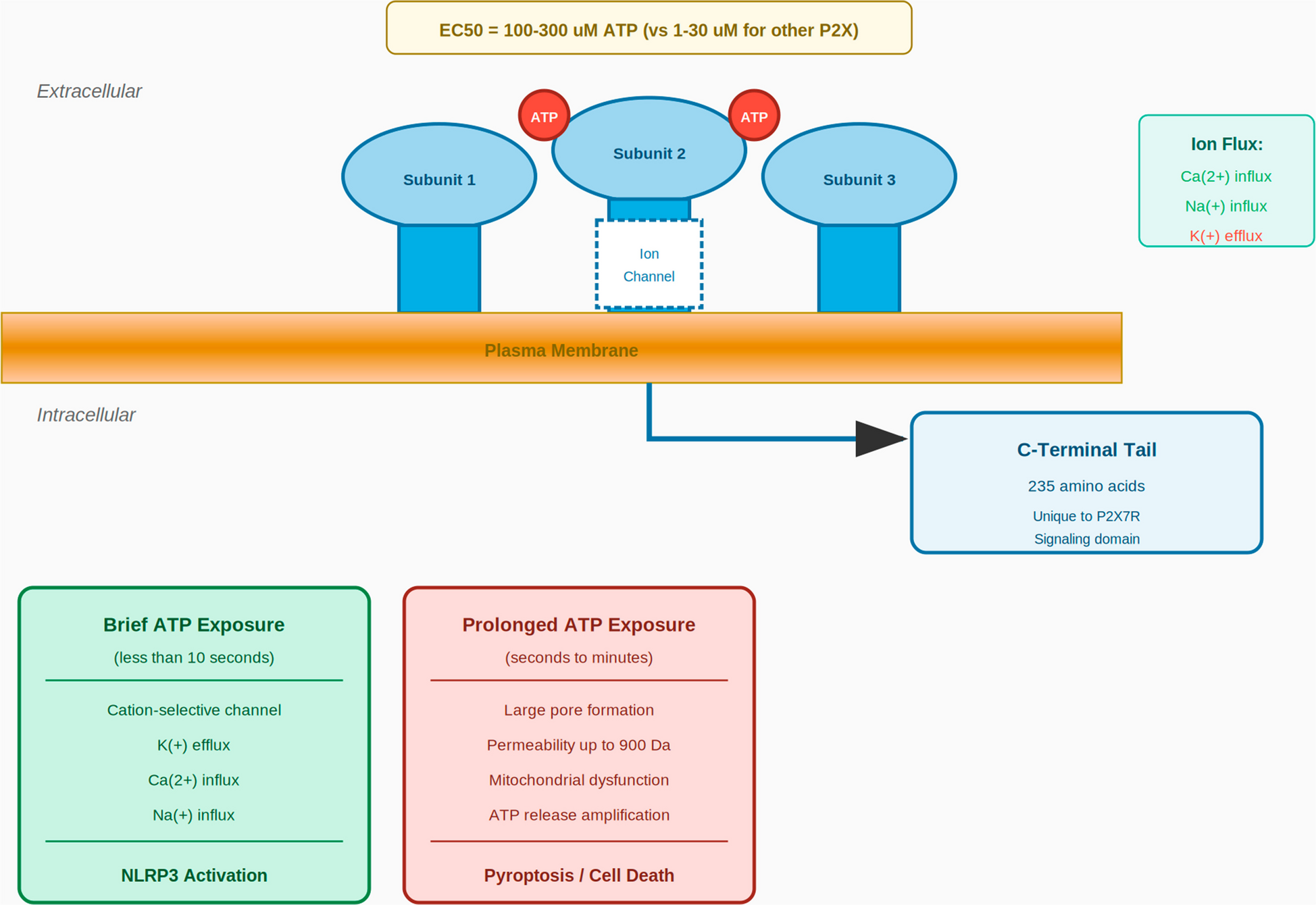
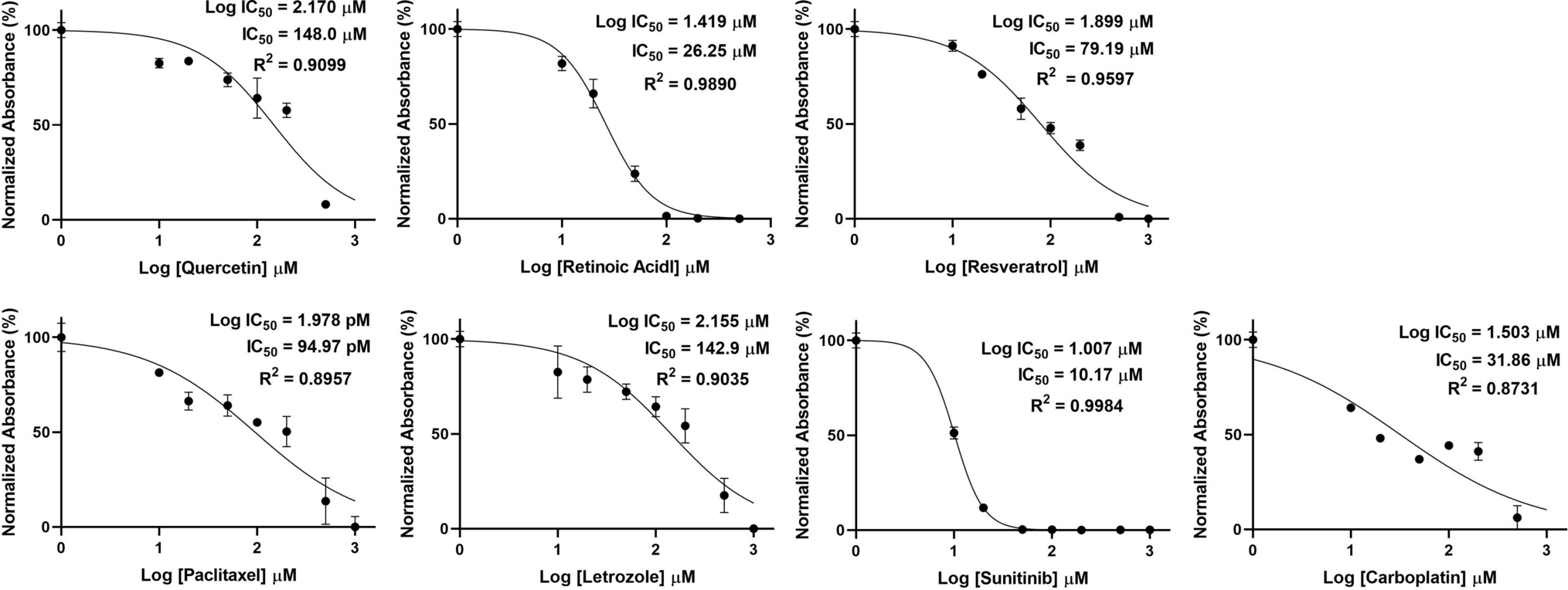
Comments (0)