Obesity is a complex chronic disease with a multifactorial aetiology that results in a pathological deposition of adipose tissue [1]. In recent decades, there has been an increasing focus on the study of disruptions in several domains of cognitive function as a potential contributing factor to the perpetuation of maladaptive eating behaviours.
Previous evidence from neuropsychological studies suggests the presence of impairments in neurocognitive functions in individuals with eating disorders (ED) and obesity compared to healthy individuals. Several neurocognitive factors have been identified as critical contributors to the onset and persistence of both disorders including deficits in executive functions, impaired reward processing, and altered decision-making [2,3,4,5,6]. Investigating these neurocognitive mechanisms is essential for developing targeted therapeutic approaches that address the underlying cognitive and neural dysfunctions. Furthermore, a comprehensive understanding of these interrelations offers promising avenues for improving diagnosis, prevention, and treatment strategies that transcend traditional categorical distinctions between obesity and ED.
This article aims to synthesize current evidence on the neurocognitive factors implicated in obesity and ED, underscoring the importance of integrated research and clinical approaches to address their comorbidity. Additionally, it seeks to empirically examine the association between neurocognitive measurements and appetite regulating hormones in two clinical groups: women with obesity, and women with obesity and comorbid ED.
1.1 Neurocognitive factors in obesity
In the last decades, a growing number of studies have demonstrated a robust association between obesity and significant impairments across various domains of neurocognitive functions, particularly in executive functions, attentional processes, and memory performance [2]. Converging neuroimaging studies corroborate these behavioural findings, revealing that obesity negatively affects the brain structure and function in regions implicated in cognitive control, including the prefrontal cortex, hippocampus and fronto-mesolimbic circuitry; as well as altered cortical morphometry [7]. In consequence, individuals with obesity show abnormal responses to food cues [7, 8]. Although the underlying mechanisms of this relationship remain unclear, increasing evidence from both human and animal studies suggests that these neurocognitive deficits are driven by a multifactorial pathophysiological process involving obesity-related insulin resistance, oxidative stress, blood–brain barrier (BBB) alterations, and neuroinflammation, which together trigger a cascade of changes that ultimately lead to synaptic remodelling and compromise the integrity of critical neural pathways [6, 9].
1.1.1 Cognitive functions in obesity: Executive functions, attention, and memory
Cognitive functions encompass a range of mental processes involved in the acquisition, processing, and utilization of information, including attention, memory, language, perception, and problem-solving [10]. Within this domain, executive functions refer to a set of top-down cognitive control processes that facilitate goal-directed behaviour, such as planning, inhibitory control, cognitive flexibility, and working memory. These functions are primarily mediated by the prefrontal cortex (PFC), which plays a central role in regulating thoughts and actions in accordance with internal goals, particularly in situations that require the suppression of salient external cues and the integration of information for complex reasoning [11].
Obesity has been consistently associated with cognitive decline across several domains, particularly executive functions, attention, and memory, suggesting a complex interplay between physiological and neurocognitive processes. This association has prompted the development of several hypotheses proposing that such impairments may not only result from obesity but also actively contribute to its development and maintenance [12].
Excess body fat has been proposed as a factor contributing to cognitive alterations in obesity. Increases in body mass index (BMI) have been specifically correlated with declines in overall cognitive performance [13]. Other indices of fat distribution, such as the weight-adjusted waist index, which reflects visceral adiposity, show a non-linear negative correlation with cognitive functioning, with the strongest associations observed in learning, memory, verbal fluency, and processing speed, particularly among older adults [14]. Taken together, higher levels of body fat have been consistently linked to impairments in verbal fluency, visual pattern memory, working memory, and executive functions, especially cognitive flexibility, suggesting that excess fat may contribute to domain-specific neurocognitive decline [14,15,16,17].
Beyond being a potential consequence of obesity and dietary factors, cognitive impairments can also contribute to maladaptive eating behaviours that promote weight gain. Individuals with obesity have been shown to exhibit memory impairments, particularly in episodic and working memory [18], which could impede the correct processing of information necessary for the self-regulation of eating behaviour [19], and may contribute to reducing adherence to nutritional guidelines and ultimately affecting treatment outcomes. Furthermore, impairments in inhibitory control could facilitate compulsive or impulsive food intake [20]. Regarding attention, individuals with obesity present reduced sustained attention and difficulties in attentional control, which may enhance vulnerability to environmental food cues. This heightened sensitivity can generate attentional biases toward highly palatable food stimuli, reinforcing maladaptive eating patterns [21, 22].
1.1.2 Dietary influences in cognitive impairment
In rodent models, experimental studies demonstrated that high-fat diets can act as pathological triggers by promoting neuroinflammation, which in turn impairs learning and memory [23]. Such effects are particularly pronounced during aging, where heightened neuroinflammation, reflected in increased microglial activation, contributes to hippocampal and amygdala damage and consequent long-term memory deficits [24]. Classic studies by Greenwood and colleagues [25,26,27,28] consistently showed that diets rich in saturated fats impair hippocampal-dependent tasks, likely mediated by metabolic disturbances such as insulin resistance. More recent work has expanded these findings, revealing that exposure to free-choice high-fat, high-sugar diets induces learning deficits accompanied by alterations in the hippocampal and cortical kynurenine pathway, again suggesting a neuroinflammatory mechanism [29]. Similarly, aged rats consuming high-calorie diets exhibit compromised episodic memory and reduced brain-derived neurotrophic factor (BDNF) and synaptic plasticity markers, linking dietary excess to neural degeneration and general cognitive decline [30].
In humans, dietary composition has also been associated with structural and functional brain changes. Diets rich in saturated fats, refined carbohydrates, and energy-dense foods, often combined with overeating behaviours, have been linked to reduced hippocampal volumes [31]. In particular, the consumption of ultra-processed foods (UPFs) has been consistently associated with overweight, obesity, and overall adverse health outcomes [32, 33]. Cross-sectional evidence indicates that higher UPF intake is related to worse verbal fluency in older adults without pre-existing health conditions [34], while longitudinal studies suggest that elevated UPF consumption is associated with accelerated decline in executive functions and an increased risk of dementia spectrum disorders, potentially mediated by a greater incidence of cerebrovascular lesions [35, 36].
1.1.3 Reward system dysregulation and impulsivity in obesity: Neuroanatomical and functional perspectives
Studies on functional brain connectivity suggest that obesity is associated with atypical architecture across prefrontal, sensorimotor cortex, insular and default mode circuits. These alterations may impair the integration of internal and external signals, leading to uncontrolled eating and excessive hedonic consumption [37, 38].
Obesity is increasingly recognised as a condition involving significant dysregulation of the brain’s reward system (RS), implying complex interactions between cognitive processes and neural circuits underlying this system [37]. At the cognitive and behavioural level, the RS supports reinforcement learning and decision-making by assigning value to stimuli, shaping motivation, and guiding goal-directed behaviour. Neuroanatomically, it is organised around the mesolimbic dopaminergic pathway and includes the nucleus accumbens, amygdala, ventral tegmental area, PFC, and hippocampus, which together mediate behaviours associated with pleasure and motivation. Dysregulations commonly observed in obesity partly resemble those in substance-use disorders, marked by heightened reactivity to specific cues, deficits in inhibitory control, and dopaminergic alterations that distort bias reward perception and pursuit. When inhibitory control is weakened, the PFC fails to regulate dopamine-driven urges, thereby compromising control over craving and impulsive decisions [39, 40].
Neuroimaging studies further indicate that in obesity, heightened responsivity to food-related cues translates into increased activation of the mesolimbic dopaminergic pathway, accompanied by altered functional and directed connectivity between prefrontal control regions and RS-related structures such as the amygdala and nucleus accumbens. These alterations suggest an imbalance between cognitive control and affective–motivational processing, leading to heightened responsiveness to food-related cues, which may in turn promote excessive hedonic consumption through less efficient top–down modulation of emotional and reward-related signals [41, 42].
1.1.4 Role of the dorsolateral prefrontal cortex (dlPFC) in cognitive control and reward regulation in obesity
Within the reward-control imbalance described above, the dlPFC plays a central role in decision-making, impulse control, and reward value processing, with its main effect on dietary behaviour being the modulation of cue-elicited cravings. Effective dietary self-regulation depends on the ability to prioritise long-term health goals, such as maintaining a balanced diet, over immediate reward-driven impulses, a process that relies critically on dlPFC-mediated top-down control [43]. Obesity is associated with heightened activation of the dlPFC and posterior parietal cortex, as well as aberrant functional connectivity within the executive control network and its interactions with the RS, linking these anatomical and functional alterations to higher impulsivity and weaker cognitive control [44].
1.1.5 Other key components of the reward system in obesity
Considering other regions functionally related to the RS, functional Magnetic Resonance Imaging (fMRI) studies have shown increased functional connectivity in individuals with obesity, particularly between the ventral striatum and the somatosensory cortex [45]. Moreover, individuals with obesity exhibit reduced dopamine D2 receptor availability in the striatum, which is associated with diminished responsiveness to palatable food intake [46, 47].
In this line, findings from fMRI showed that individuals who gained weight displayed reduced striatal responses to palatable food cues compared to those who maintained their weight. This suggests that as weight increases, the RS becomes less sensitive to appetitive stimuli. The reduced striatal responsivity could represent a mechanism driving overeating: when food is perceived as less rewarding at the neural level, individuals may increase consumption in an attempt to compensate for this ‘reward deficit’, thereby promoting further weight gain. This pattern mirrors tolerance mechanisms observed in substance addiction [47].
Taken together, these findings underscore the broader architecture of the RS as key neural substrates for self-regulation processes in obesity.
1.2 Effects of obesity-related disorders on brain structure and function1.2.1 Insulin resistance and cognitive function: How insulin resistance impact brain function
The presence of insulin-related multimorbidity has been demonstrated to be associated with an increased risk of cognitive impairment [48]. Evidence has emerged linking the presence of type 2 diabetes and obesity-related insulin resistance with varying stages of cognitive decline, ranging from mild cognitive impairment to dementia [49,50,51].
As has been previously documented, although glucose can enter the central nervous system (CNS) by diffusing across BBB, insulin receptors are expressed on all cell types in the brain, which suggests that it plays diverse critical roles in the brain [51] regulating glucose metabolism, synaptic plasticity, and neurotransmission release [52]. Such functions have been particularly observed in regions of the hippocampus and PFC [53]. Levels of insulin in cerebrospinal fluid are significantly lower than those found in plasma. However, these levels are correlated, suggesting that the majority of insulin in the brain originates from circulating pancreatic insulin [51]. In the context of obesity, peripheral insulin levels corresponding to the state of insulin resistance have been suggested to correlate with those corresponding to central insulin resistance [52]. This could potentially result in a disruption to the insulin signalling within the brain, which may be associated with impaired memory formation, executive functions, and attention [52]. Central insulin resistance has been linked to hippocampal atrophy and reduced functional connectivity in the default mode network. This is a network of interacting brain regions that is highly active during rest periods, when the individual is not focused on a particular task [52]. Furthermore, the available evidence suggests that an increase in metabolic disruptions results in an aggravation of neurocognitive impairment. The presence of metabolic syndrome has been demonstrated to exacerbate neural dysfunction through mechanisms of oxidative stress and vascular damage, which increases the risk of cognitive decline and neurodegenerative diseases, including Alzheimer’s disease [51].
1.2.2 The role of leptin on brain function: Its potential impact on neurocognitive processes
Leptin, a hormone primarily secreted by adipocytes, regulates energy homeostasis by signalling satiety to the hypothalamus [54]. Beyond its role in appetite regulation, leptin has been demonstrated to influence brain regions involved in cognitive processes, including the hippocampus and PFC [55].
In individuals with obesity, leptin is overexpressed at the gene level in the adipose tissue, resulting in elevated leptin levels and impair leptin signalling pathways [56]. This pathologically increased circulating levels have been recognised by some authors as a biomarker of leptin resistance [57]. This is defined by a reduced sensitivity or a failure in response, which results in a decreased ability to suppress appetite or enhance energy expenditure. Ultimately, this leads to overeating and subsequent weight gain [57]. In addition, preclinical studies have shown that these elevated levels also result in a combination of a decreased BBB transport of leptin, and reduced in leptin receptor signalling. This disrupts hippocampal synaptic plasticity, which is essential for memory formation; and increases cognitive deficits and the risk of neuropsychiatric disorders [58, 59].
1.2.3 Chronic inflammation and brain function: The Role of Inflammatory Markers in Neuroinflammation and Cognitive Impairments in Obesity
Obesity has an important inflammatory component. It is characterised by altered serum levels of inflammatory cytokines such as Tumour Necrosis Factor-alpha (TNF-α), C-reactive protein (CRP) and interleukins (IL-6, IL-18), among others [60]. These inflammatory mediators can access the brain and activate the microglia, brain’s immune cells, which further release locally cytokines and reactive oxygen species (ROS), leading to neuroinflammation [61]. This local inflammatory state has a negative impact on synaptic plasticity, particularly in the hippocampus and PFC [62].
Clinical studies have found a correlation between higher levels of serum inflammatory markers and poorer cognitive performance in adults with obesity [61]. A prospective study with the objective of evaluating low-grade inflammation and a decline in cognitive performance over a follow-up period of 10 years in adults with overweight identified TNF-α and IL-6 as predictors of cognitive decline. This finding suggests that low-grade inflammation was associated with poorer cognitive performance in later life [63]. In this line, the results of a study conducted on a young adult cohort with obesity showed that higher CRP concentrations had a significant negative association with poorer neuropsychological performances of visuospatial attention [64].
Furthermore, obesity-related inflammation can alter neurotransmitter systems, particularly those pathways involving dopamine and serotonin, which play a crucial role in regulating mood and cognition [65]. Reduced dopamine synthesis induced by inflammation has been linked to anhedonia and cognitive impairment, suggesting a mechanistic link between obesity, inflammation and neuropsychiatric symptoms [66].
1.2.4 Gut-brain axis: Gut microbiome and brain function in obesity
The term gut-brain axis is used to describe the bidirectional communication network involving neural, endocrine and immune pathways that connect the gastrointestinal tract and the CNS. The gut microbiota plays a vital role in this axis through the modulation of metabolic function, immune response, and neurotransmitter production [67].
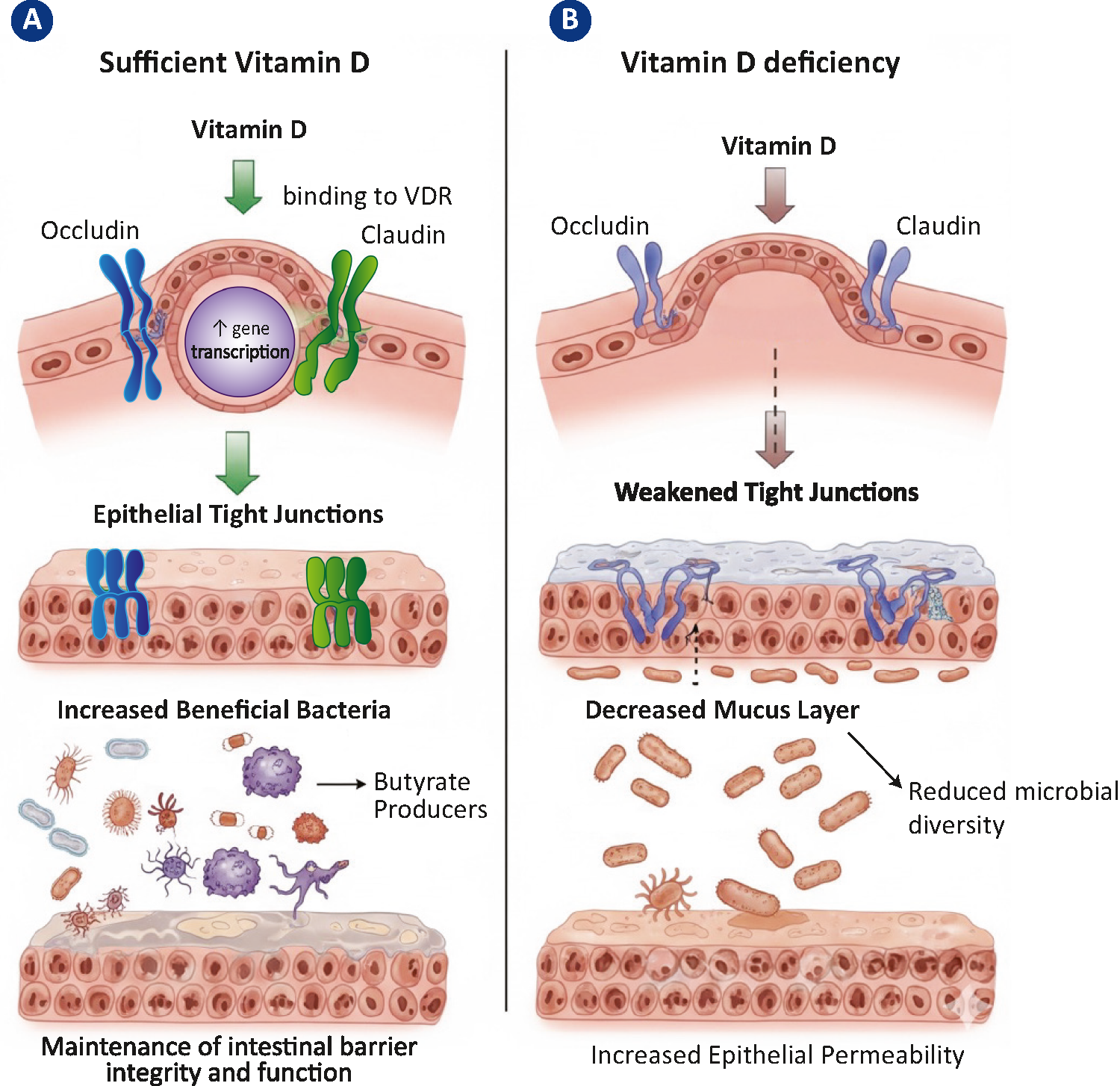
The presence of obesity has been linked to gut dysbiosis, a condition characterised by a reduction in microbial diversity and an alteration in bacterial populations [68]. Dysbiosis has been demonstrated to resulting diminished production of beneficial metabolites, including short-chain fatty acids (SCFAs), especially butyrate. In addition, these SCFAs have been shown to possess both anti-inflammatory and neuroprotective properties [68, 69]. Suc

Comments (0)