
Remember me
This study was conducted as a pharmacokinetic sub-study within the V-SMART trial (ACTRN12620000681954) to evaluate the feasibility of saliva sampling for TDM of linezolid in patients with multidrug-resistant tuberculosis (MDR-TB). The study was prospective, observational, and conducted across four provinces in Vietnam. Ethics approval was obtained from the University of Sydney, Australia (Protocol No. 2021/082, approved on 21 October 2021) and the National Lung Hospital, Vietnam (Approval No. 25/21/CN-HDDD, dated 27 April 2021).
Adults aged ≥ 18 years who had received linezolid treatment for ≥ 7 days (to ensure steady-state conditions) and had consented to multiple blood and saliva collections over a 5-hour sampling window (pre-dose, 2- and 5-h post-dose) were eligible. Individuals unable to provide consent or with oral conditions interfering with saliva collection were excluded. All participants provided written informed consent prior to enrolment.
2.2 Quantification of Linezolid in Plasma and SalivaLinezolid concentrations in plasma and saliva were quantified using a validated LC-MS/MS method [21]. Briefly, blood samples were collected in EDTA tubes and centrifuged, whilst saliva samples were collected in Salivette® tubes. The saliva samples were filtered through a 0.22 µm Millex-GP filter to disinfect the M. tuberculosis [22]. After filtration, all samples were aliquoted, frozen and stored at − 80 °C until analysis. Samples preparation included protein precipitation with methanol containing deuterated internal standards to remove particles that could interfere with the assay. Quantification was performed using a Quadrupole-Orbitrap mass spectrometer following chromatographic separation. Plasma and saliva concentrations that were below the assay’s lower limit of quantification (LLOQ = 0.5 mg/L) were treated as missing observations and excluded from the analysis (Beal’s M1 method). Unbound concentrations were not measured because this requires specialised methods rarely available in low-resource settings, so would not be implementable [23]. Total plasma concentrations correlate well with unbound plasma and saliva concentrations for linezolid (r ≈ 0.9, p < 0.05) [24], making them reasonable surrogates for this analysis.
2.3 Population Pharmacokinetic AnalysisThe PopPK analysis of linezolid was performed using nonlinear mixed-effect modelling in NONMEM (version 7.4.4; ICON Development Solutions, Hanover, MD, USA) with first-order conditional estimation and eta-epsilon interaction (FOCE-I) and ADVAN13 subroutine. Data visualisation and post-processing were conducted in R (version 4.3.3; R Foundation for Statistical Computing, Vienna, Austria). Model development was supported using Pirana (version 21.11.1; Certara, Princeton, NJ, USA), Perl-Speaks-NONMEM (version 5.3.1) [25], and Xpose4 [26].
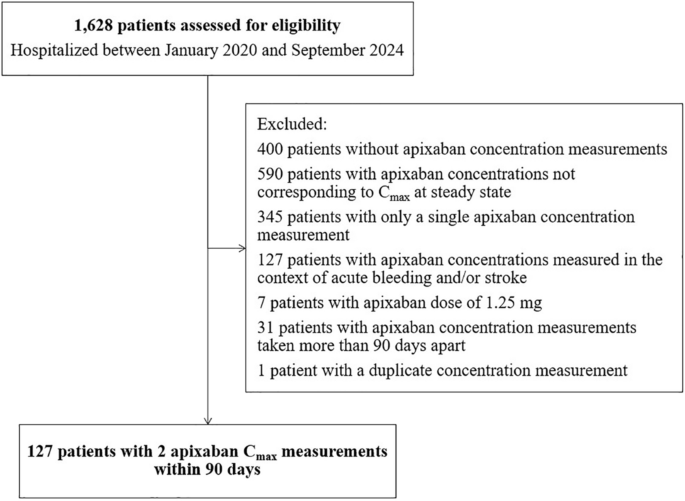
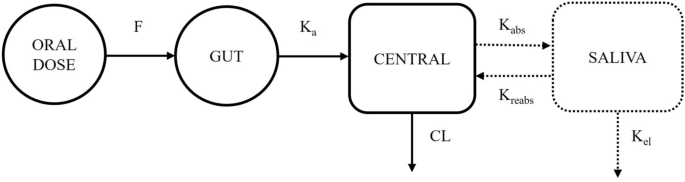
A stepwise approach was used to develop combined plasma-saliva pharmacokinetic model. Plasma concentrations were first modelled using one- and two-compartment structures with first-order absorption and linear elimination (Supplementary Information S1). A literature-informed one-compartment model served as the starting point for model development [27]. Interindividual variability (IIV) was assessed on structural parameters based on η-shrinkage, model stability, and successful completion of the covariance step. Residual unexplained variability (hereafter referred to as residual error) was tested using additive, proportional, and combined error structures. Nested models were evaluated using the likelihood ratio test, while non-nested models used Akaike information criterion. Model performance was assessed through diagnostic plots and visual predictive checks. Subsequently, saliva data were added by evaluating either a separate saliva compartment or scaling from the central compartment. Model structures were compared graphically (Fig. 1 for the distinct saliva model and Supplementary Figure S1 for the scaled saliva model) [28, 29]. Residual error in saliva concentrations was modelled separately.
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.The conceptual model for linezolid PK in saliva and plasma. The solid and dotted lines represent linezolid PK in plasma and saliva, respectively. CL central clearance, F oral bioavailability, Ka absorption rate constant, Kabs first-order absorption rate of saliva, Kel salivary elimination rate, Kreabs first-order reabsorption rate of central compartment, PK pharmacokinetic. The saliva bio-compartment is a hypothetical effect compartment
Covariate analysis explored associations between pharmacokinetic parameters and covariates (e.g. body weight, age, renal, and hepatic function markers). Exploratory plots guided variable selection, followed by stepwise covariate modelling (forward inclusion: ∆–2log-likelihood ≥ 3.84, p < 0.05; backward elimination: ∆–2log-likelihood ≥ 6.63, p < 0.01). Sex was not assessed due to limited representation (14 males vs 3 females). Continuous covariates were modelled using a linear or power function (Supplementary Information S2).
Model evaluation included parameter precision, reduction in objective function, successful covariance/correlation steps, and goodness-of-fit diagnostics. Final parameter uncertainty was assessed via sampling importance resampling (SIR) in Perl-Speaks-NONMEM [30], which was adopted to acquire 95% confidence intervals (CIs) and to evaluate the precision of the final model estimates. A samples/resamples (M/m) ratio of 5000/1000 was used [30]. The predictive performance of the final model was internally validated using visual predictive check with 1000 replicates [31].
2.4 Simulation of Limited Sampling StrategiesModel applicability was examined through two simulation scenarios. First, assessing whether the current saliva sampling schedule (pre-dose, 2-, and 5-h post-dose) accurately predicts plasma AUC0–24 of linezolid (Supplementary Figure S2 for a visual demonstration). Second, exploring alternative saliva-based sampling strategies were investigated to determine optimal timepoints for accurate plasma AUC0–24 estimation (Supplementary Figure S3).
2.4.1 Predictive Performance of the Current Saliva Sampling StrategyPlasma AUC0–24 reference values were derived by reconstructing full 25-point plasma profiles (sampled hourly from 0 to 24 h) from observed plasma data (pre-dose, 2, 5 h post-dose) via Bayesian maximum a posteriori (MAP) estimation using the final popPK model. The Perl-Speaks-NONMEM 'keep_estimation' option and MAXEVAL = 0 ensured predictions were based on individual post hoc parameters. Unmeasured timepoints were imputed.
To incorporate IIV variability, 300 Monte Carlo replicates were per subject (N = 17) generated 5100 virtual subjects. Simulations were based on the final popPK model, maintaining the original study design and covariate relationships, while integrating the variance-covariance matrix to preserve correlations among pharmacokinetic parameters. Reference plasma AUC0–24 values were then calculated from simulated profiles using the linear trapezoidal rule implemented in noncompartmental analysis posterior predictive checks package. To evaluate saliva-based prediction, the same process was applied using saliva data (pre-dose, 2, 5 h post-dose) as Bayesian priors. Predicted plasma profiles were reconstructed, and AUC0–24 was derived. Agreement between saliva-based predicted with reference plasma AUC0–24 values was assessed using Wilcoxon signed-rank tests (α = 0.05). To further illustrate the magnitude and direction of discrepancies between the two approaches, Passing-Bablok regression and Bland-Altman analysis were also performed. Clinical acceptability was defined as predicted AUC0–24 within ± 20% of reference values [32, 33].
2.4.2 Alternative Saliva Sampling StrategiesMultiple limited sampling strategies were systematically assessed to identify saliva timepoints capable of accurately predicting plasma AUC0–24. Sampling strategies consisting of one, two, or three saliva samples were evaluated. Potential sampling timepoints were selected based on linezolid’s pharmacokinetic profile and clinical feasibility, and included pre-dose, 2, 5, 6, 8, 10, 12, 14, and 16 hours after dosing. Because only three saliva concentrations (pre-dose, 2, and 5 h post-dose) were available, additional timepoints were imputed using Bayesian MAP estimation based on individual pharmacokinetic profiles.
In the one-sample saliva strategy, six cases were assessed, with saliva samples collected at either pre-dose, 2, 5, 8, 10, or 12 h post-dose. When the selected saliva timepoint corresponded to an observed value (i.e., pre-dose, 2 or 5 h), the missing dependent variable was coded as zero at the point and one at all unobserved plasma timepoints. For imputed timepoints (8, 10, or 12 h post-dose), individual predicted concentrations were computed via Bayesian MAP estimation and treated as pseudo-observations by assigning a missing dependent variable value of 0 at the selected timepoint and 1 at all others.
For the two-sample strategies, six combinations were explored: (2 and 5 h post-dose), (2 and 6 h), (2 and 8 h), (2 and 10 h), (2 and 12 h), and (2 and 14 h). Each scenario generated a dataset containing two saliva concentrations per subject. Six three-sample strategies were also examined: (pre-dose, 2, 6 h post-dose), (2, 5, 8 h), (2, 5, 10 h), (2, 5, 12 h), (2, 5, 14 h), and (2, 5, 16 h), with each combination resulting in a dataset that included three saliva measurements per subject.
Each of the 18 saliva sampling strategies was simulated using 300 Monte Carlo replicates per subject (N = 17), generating 5100 virtual profiles. Plasma AUC0–24 values were derived from the simulated plasma concentration–time profiles using the linear trapezoidal rule implemented in the noncompartmental analysis posterior predictive checks framework. To assess predictive accuracy, saliva-based AUC0–24 estimates were compared with reference AUC0–24 values (derived using the current pre-dose, 2, 5 h post-dose plasma sampling strategy) using the Wilcoxon signed-rank test (α = 0.05) and regression analyses. A pre-defined threshold ±20% deviation from the reference AUC0–24 was used to determine clinical acceptability [32, 33].
To assess the clinical interpretation of plasma- and saliva-based TDM, the efficacy of linezolid was defined as fAUC0–24/MIC of ≥ 125 [9, 10]. The unbound fraction of linezolid was assumed to be 87.2% [34] and a surrogate MIC of 0.5 mg/L was used to reflect common M. tuberculosis strains [35]. Because linezolid tablets are available only in 400-mg and 600-mg strengths (with some 600-mg tablets scored), the smallest practical dose adjustments are 300 mg. This constraint was considered when interpreting the clinical impact of predicted exposure differences. For each patient, we calculated the proportion and magnitude of discordant interpretations between plasma- and saliva-based TDM. Monte Carlo simulations (5100 virtual patients per scenario) based on the final popPK model were performed to estimate how frequent saliva-based AUC0–24 estimates would lead to different dose recommendations than plasma-based AUC0–24 estimates. Simulations evaluated multiple sampling strategies (trough and the pre-dose, 2 and 6 h post-dose strategies) and for iterative saliva-based plasma predictions (1, 2, or 5 samples). For each scenario, saliva-based and reference plasma AUC0-24 estimates were compared, and disagreements were classified by direction (efficacy- vs toxicity-driven) and by magnitude of the implied dose change (< 20%, 20–50%, 50–100%, > 100%).
Comments (0)