
Remember me
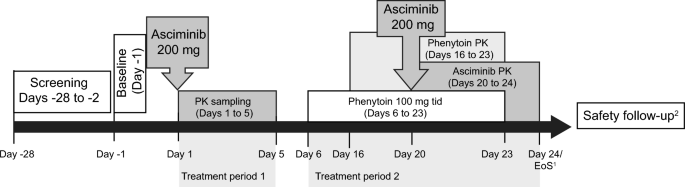
This was a Phase 1, open-label, fixed-sequence, DDI study consisting of a screening period, two treatment periods, and a safety follow-up period of 30 days after the last dose of asciminib (Fig. 1).
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Study design. 1EoS defined as the date of the last scheduled procedure. 2A post-study safety contact (e.g., follow-up telephone call or email) occurred approximately 30 days after the last administration of study treatment. EoS end of study, PK pharmacokinetics, tid three times daily
During treatment period 1 (Days 1–5), participants received a single dose of asciminib 200 mg on Day 1 after an overnight fast (for at least 10 h). Participants continued to fast for an additional 4 h post-dose. A 5-day washout period was scheduled between the first dose of asciminib and the first dose of phenytoin. During treatment period 2 (Days 6–24), participants received phenytoin 100 mg three times daily (tid) from Days 6–23, administered approximately 8 h apart (±1 h). On Day 20, the morning dose of phenytoin and asciminib was co-administered under fasted conditions (10 h before dosing), and participants continued to fast for an additional 4 h post-dose (as done on Day 1). The absorption of asciminib is affected by food and therefore needs to be taken in the fasted state at all doses [9, 10]. The phenytoin dosing preceded asciminib dosing by 14 days and continued for 3 days after asciminib dosing to ensure continued maximal CYP3A4 induction through the majority of the asciminib systemic exposure time course.
Participants were confined during the in-clinic study treatment period for up to 25 days and 24 nights. Asciminib and phenytoin were taken orally with approximately 240 mL of water. Additional water was permitted ad lib, except for 1 h before and until 1 h after asciminib administration on Days 1 and 20. No medication other than the study treatment was allowed from the date the participants signed the informed consent until completion of the end-of-study evaluations, except for protocol-allowed concomitant medication and/or any medication that could be required to treat adverse events (AEs).
2.2 ParticipantsEligible participants were healthy male or female (non-childbearing potential) adults (aged 18–55 years) in good health as determined by no clinically significant findings from medical history, physical examination, vital signs, and laboratory data, and no significant findings from electrocardiogram (ECG) data or electrolytes in blood or urine, including findings not considered clinically significant. Other inclusion criteria were body mass index (BMI) ranging from 18.5 to 29.9 kg/m2, body temperature of 36.0 to 37.5 °C, systolic blood pressure of 100 to 140 mmHg, diastolic blood pressure of 70 to 90 mmHg, and pulse rate ranging from 60 to 90 bpm.
Key exclusion criteria were a history of any clinically significant medical conditions, treatment with investigational drugs within 30 days or longer if required by local regulation (or five half-lives, whichever is longer) before study dosing and contraindications, hypersensitivity, or any previous adverse reactions to the compound, compound class, or excipients of asciminib or phenytoin or other hydantoins (including ethotoin, mephenytoin, and fosphenytoin).
2.3 Objectives and EndpointsThe primary objective of the study was to evaluate the effect of phenytoin as a strong inducer of CYP3A4 on the PK of asciminib following a single oral dose of 200 mg in healthy participants. Primary endpoints were maximum (peak) observed plasma drug concentration following dose administration (Cmax), area under the curve (AUC) from time zero to the last measurable plasma concentration sampling time (AUClast), and AUC from zero to infinity (AUCinf) of asciminib when administered with phenytoin versus asciminib alone. Secondary plasma PK parameters included time to maximum observed plasma drug concentration (Tmax), apparent terminal elimination half-life (T1/2), AUC from time zero to time t (AUC0-t), time that corresponded to the last measurable concentration (Tlast), total apparent body clearance (CL/F), and apparent volume of distribution during the terminal phase (Vz/F) for asciminib.
Secondary objectives were to assess the safety and tolerability of a single oral dose of asciminib 200 mg given alone or concomitantly with phenytoin in healthy participants and to assess trough concentrations of phenytoin. Secondary endpoints were safety endpoints (including vital signs, physical examinations, ECG findings, safety laboratory parameters, AEs coded using Medical Dictionary for Regulatory Activities version 27 (MedDRA) and Common Terminology Criteria for AEs version 5.0), and phenytoin plasma Ctrough levels.
Moreover, the effect of a single oral dose of asciminib 200 mg on CP-1, which is an endogenous OATP1B substrate, was assessed as an exploratory endpoint. Coproporphyrin-1 PK parameters were the plasma pre-dose concentration, Cmax, and AUC from time zero to 24 hours (AUC0-24).
2.4 PK Sampling and AssessmentsFor the assessment of asciminib plasma concentrations, serial blood samples were collected following asciminib administration on Day 1 (treatment period 1) and Day 20 (treatment period 2): pre-dose (0 h) and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 72, and 96 h. Pharmacokinetic samples for phenytoin quantitation were collected from Days 16 to 23 before the morning dose. During treatment period 1, CP-1 PK plasma sampling was performed on Day 1 at pre-dose and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h post-dose. Plasma concentrations of asciminib, phenytoin, and CP-1 were determined using validated liquid chromatography-tandem mass spectrometry assays, with the lower limit of quantification values of 1.0, 60.0, and 0.05 ng/mL, respectively. The PK parameters were determined from plasma concentration-time data using noncompartmental methods employing Phoenix WinNonlin software, version 8.3, using the actual sampling times.
2.5 Sample Size and Statistical AnalysisThe sample size was chosen based on practical considerations and to obtain a desired precision of the 90% confidence interval (CI) around the estimated geometric mean ratio. A conservative intra-coefficient of variation (CV) of 25% was derived from the maximum intra-subject CV% for AUClast, AUCinf, and Cmax from a previous study of asciminib administered with rifampicin [12]. With a sample size of 12 participants, the precision or half width of the 90% CIs for test-reference comparison on the log scale was calculated to extend 0.181 from the observed difference in means. Considering a potential drop-out rate of 20%, a target of 15 participants was selected.
The safety analysis set, which included all participants who received at least one dose of any study treatment (asciminib or phenytoin), was used for safety evaluations. The PK analysis set (PAS) for asciminib (PAS-asciminib) included all participants who provided an evaluable asciminib PK profile for at least one period. The PAS for phenytoin (PAS-phenytoin) included all participants who provided at least one evaluable phenytoin Ctrough value in treatment period 2.
If a participant did not meet the PAS definition, all scheduled concentrations for that participant as well as corresponding parameters were excluded from analyses. Additionally, if adjusted R2 was <0.75 or the percentage extrapolated AUC was >20%, then λ₋z, T1/2, AUCinf, Vz/F, and CL/F were excluded from analyses. Missing values for any PK parameters or concentrations were not imputed and were treated as missing. Drug concentrations below the lower limit of quantitation (LLOQ) were set to zero by the bio-analyst. Concentrations below LLOQ were treated as missing for the calculation of the geometric means and geometric CV%, and as zero for all other calculations including calculation of PK parameters. Participants with missing PK parameters (e.g., AUCinf, AUClast, Cmax) in one but not both periods were included in the mixed model analysis assuming missing at random.
For asciminib Cmax, AUClast, and AUCinf, a linear mixed-effects model was fitted to the log-transformed PK parameters to estimate the effect of phenytoin at steady-state on the PK of a single-dose of asciminib 200 mg. The model included treatment as a fixed factor and participant as a random factor. For each of the PK parameters, a point estimate and the corresponding two-sided 90% CI for the difference between the means of the test (asciminib 200 mg + phenytoin 100 mg tid) and reference (asciminib 200 mg) treatments were calculated. The means were anti-log transformed to obtain the point estimate and the 90% CI for the geometric mean ratio on the original scale. The linear trapezoidal rule was used for all AUC calculations. For asciminib, regression analysis of the terminal plasma elimination phase for the determination of T1/2 included at least 3 data points after Cmax. All statistical analyses were performed using Statistical Analysis System (SAS), version 9.4.
Comments (0)