
Remember me

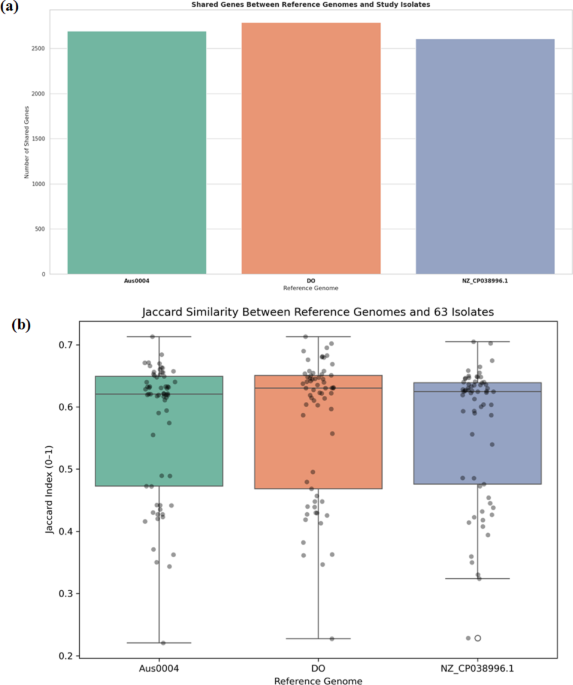

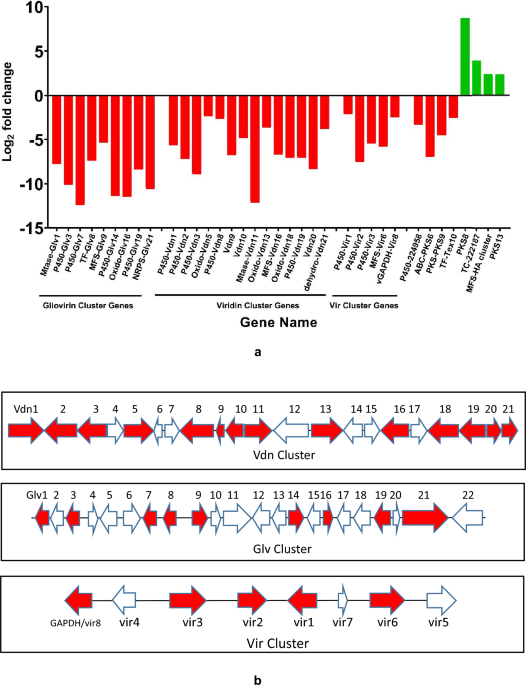
To identify the most suitable reference genome for downstream comparative analyses, three E. faecium reference genomes, Aus0004, DO, and SRR24 were evaluated using gene presence/absence matrix obtained from Roary. Among the three reference genomes analyzed, E. faecium DO showed the highest similarity to the clinical isolates based on pan-genome clustering and shared gene content (Fig. 1a). It displayed a moderate number of accessory genes (n = 451), suggesting a balance between genomic conservation and variability. A Kruskal-Wallis test revealed a statistically significant difference in accessory gene count among these references (H = 14.0, p = 0.00091), suggesting variability in genome plasticity. To further assess genome similarity, Jaccard similarity index was calculated between each reference genome and all 63 study isolates based on shared gene content. The mean ± SD Jaccard indices were: DO (0.577 ± 0.112), Aus0004 (0.571 ± 0.110), and SRR24 (0.565 ± 0.110) (Fig. 1b). However, the Jaccard similarity index test revealed no statistically significant difference across the three groups (H = 1.581, p = 0.45356), indicating comparable levels of overall gene similarity between the references and the isolates. Based on the combination of moderate accessory gene content and the highest average Jaccard similarity, the DO genome was selected as the representative reference for subsequent SNP-based and comparative genomic analyses.
Pangenome analysis of the E. faecium isolates revealed a total of 29,196 genes, which were classified based on their frequency of occurrence in the analyzed strains. The core genome, which includes genes found in 99% to 100% of isolates, contained 1,660 genes, reflecting the fundamental conserved genetic framework for the species. The soft-core genome, comprising genes found in 95% to less than 99% of the strains, included 266 genes. The shell genome, which represents moderately conserved genes present in 15% to less than 95% of strains, accounted for 1,987 genes (Fig. 2a). The largest component of the pangenome was formed by the cloud genes, which were rare and found in less than 15% of the strains, totaling 25,283 genes. This distribution highlights a substantial genetic diversity within the population, with a relatively small conserved core and a large proportion of accessory genes. The gene presence/absence matrix generated by Roary was visualized alongside a dendrogram based on gene content similarity among all E. faecium isolates (Fig. 2b). The matrix displays a binary heatmap where each row represents a genome and each column corresponds to a gene cluster (n = 29,196). Dark blue blocks indicate the presence of specific gene clusters, while light areas represent absence. The corresponding tree, constructed using the accessory gene presence-absence profiles, reveals distinct clustering patterns, suggesting gene content-driven phylogenetic relationships. Closely clustered isolates share large segments of gene content, while more distantly related strains display sparse overlap, reflecting both the diversity of the accessory genome and potential niche adaptation or horizontal gene transfer events among lineages. The frequency distribution of gene clusters based on their presence across genomes is illustrated in Fig. 2c.
Phylogenetic analysis, MLST and plasmid replicon distributionA maximum likelihood phylogenetic tree was constructed using core genome SNP alignment data from 63 Enterococcus isolates. Tree inference was carried out with IQ-TREE, and support for internal nodes was assessed using ultrafast bootstrap replicates.
To compare alternative topologies and assess the robustness of the phylogeny, an Approximately Unbiased (AU) test was performed using three candidate trees representing key reference strains: DO, AUS004, and SRR24 (S2_Supplementary File [Figure S1-S3]). The tree based on the DO reference showed the highest log-likelihood score (− 165029.17) and was strongly supported (p-AU = 0.845). In contrast, the SRR24-based tree had the lowest likelihood (− 165385.45) and was statistically rejected (p-AU = 0.0124), while the AUS0004 tree had intermediate support (log-likelihood = − 165161.22; p-AU = 0.182). These results reinforced the selection of DO as the optimal reference for downstream phylogenetic analysis. The resulting DO-based tree revealed multiple well-supported clades, indicating the presence of distinct evolutionary lineages in the clinical isolates.
Several isolate pairs or small groups, such as (Isolate_10, Isolate_30), (Isolate_12, Isolate_27), and (Isolate_34, Isolate_45), clustered with high bootstrap values (≥ 98%), reflecting close genetic relatedness. Notably, the DO reference genome clustered with Isolates 56, 60, and 62, forming a distinct and potentially divergent clade within the population, possibly representing a separate evolutionary lineage.
To investigate the population structure of E. faecium, MLST analysis identified eight distinct STs among the 63 isolates. ST80 was the most prevalent, detected in 52 isolates (82.54%), followed by ST18 (4 isolates, 6.35%) and ST17 (2 isolates, 3.17%). ST31, ST117, ST1380, ST1693, and ST1886 were each represented by a single isolate (1.59%). A SNP-based phylogenetic tree annotated with MLST profiles, year of isolation, and plasmid replicon content (Fig. 3) provided a comprehensive overview of lineage distribution and genetic relationships within the studied population.
To further examine the genetic relatedness among ST80 isolates, a pairwise SNP distance analysis was conducted. Histograms and boxplots (S2_Supplementary File [Figure S4]). revealed a wide distribution of SNP distances, ranging from around 5,000 to 8,000, with a median close to 6,300 SNPs. This wide range indicates considerable genetic diversity among ST80 isolates, implying that the prevalent occurrence of ST80 is indicative of several related yet distinct lineages, rather than a singular clonal expansion within the population.
Plasmid replicon typing was conducted on 63 E. faecium isolates to evaluate the diversity and distribution of plasmid families. A total of 32 unique replicon types were identified, revealing substantial variability in plasmid content. The isolates harbored a mix of small [e.g., pB82 (rep11), pEF418 (rep18), pCIZ2 (rep29)] and medium-to-large plasmids [e.g., rep1, rep2, pRUM (rep17), and the unique pHTβ (repUS7)]. These replicons are classified based on their rep genes, which determine plasmid replication. The most frequently detected replicon was repUS15_2_repA (pNB2354p1), followed by rep2_1_orf1 (pRE25), which were present in 10% and 9.3% of the isolates, respectively. These replicons are a well-known contributor to the mobilization of antimicrobial resistance genes. Other highly prevalent replicons included rep17_1_CDS29(pRUM) (6.4%), rep18a_1_repA(p200B) (6.2%) and rep11a_1_repA(pB82) (6.1%). The frequent detection of pRUM-like plasmids is notable, as they are often associated with vancomycin resistance in clinical strains.
In addition to these rep-typed plasmids, small Col-type plasmids replicon such as Col440I_1, Col(BS512)_1, and Col(MG828)_1 were also found in isolates. Unlike rep-typed plasmids, Col-type plasmids are characterized by their conserved Col-like replication origins and typically lack rep genes. Col-type plasmids are often mobilized by co-resident conjugative elements and often carry genes that confer resistance to antibiotics. Figure 4 illustrates the plasmid replicons identified in E. faecium isolates. In contrast, several replicons were observed at low frequencies (1–4 isolates), including rep14a_4_rep (AUS0004p3), rep14a_3_EFAU085p5001(AUS0085p5), rep14b_2_EFAU085p6001(AUS0085p6), rep29_3_ORF8(pCIZ2), rep1_1_repE(pAMβ), repUS7_2_rep(pMG1), repUS7_1_rep(pHTβ), and IncFIA_1. Overall, the plasmidome of these E. faecium isolates displayed a complex and heterogeneous structure, with many isolates harboring multiple plasmid replicon types. The high prevalence of clinically relevant replicons particularly those linked to vancomycin and aminoglycoside resistance highlights the significant role of plasmids in shaping the resistome.
Resistance gene profilingResFinder analysis identified a total of 87 distinct AMR genes across the E. faecium isolates, highlighting a highly diverse and extensive resistome with strong MDR potential. These genes spanned multiple antibiotic classes, including aminoglycosides, macrolides, glycopeptides, sulfonamides, tetracyclines, and beta-lactams.
Figure 5 illustrates the top 10 most prevalent resistance genes. The msr(C) gene was the most identified, found in almost all isolates. The msr(C) is an ATP-binding cassette (ABC) transporter which mediate resistance to erythromycin, telithromycin, and streptogramin antibiotics. The second most common resistance gene was aac(6’)-Ii associated with high-level resistance to several aminoglycosides, including gentamicin, tobramycin, amikacin, and streptomycin. erm(B) encodes an rRNA methyltransferase responsible for resistance to macrolide-lincosamide-streptogramin B (MLSB) antibiotics, including erythromycin, lincomycin, clindamycin, quinupristin, pristinamycin IA, and virginiamycin S, was also highly prevalent. The vanHAX operon was detected in 45 out of 63 isolates (71.4%), which was statistically significant compared to an expected proportion of 50% (Binomial test, p = 0.0036), indicating a high prevalence of vancomycin resistance operons among the studied isolates. This operon confers resistance to both vancomycin and teicoplanin. Other frequently identified aminoglycoside resistance genes included, aac(6’)-aph(2’’), aph(3’)-III, aph(2’’)-Ia, and ant(6)-Ia. Resistance genes targeting other antibiotic classes were also observed. These included sul1 (sulfonamides), dfrA12 (trimethoprim), tet(S) (tetracyclines, including doxycycline), as well as blaOXA-232 and blaTEM-104, which encodes resistance to beta-lactam/beta-lactamase inhibitor combinations such as ampicillin-clavulanic acid, piperacillin, and piperacillin-tazobactam. Moreover, linezolid resistance genes optrA and poxtA were detected in 14 and 2 isolates, respectively.
Several antimicrobial resistance genes, including aminoglycoside resistance genes (ant(6)-Ia, aph(3’)-III, aac(6’)-aph(2’’)), β-lactamases (blaTEM-104, blaOXA-1, blaOXA-232, blaTEM-1B), tetracycline resistance genes (tet(S), tet(L)), macrolide resistance genes (erm(B), erm(T)), the vanHAX operon, sul1, cat(pC233), and the plasmid-mediated quinolone resistance gene qnrS1, were also detected on plasmid replicons. These findings reveal that multiple plasmid replicon types co-occurred with AMR genes in several isolates, suggesting a possible contribution of plasmids to resistance gene dissemination. To further investigate the distribution of AMR genes across the isolates, a heatmap (Fig. 6) was generated. This revealed a heterogeneous pattern, with some isolates harboring a broad spectrum of resistance genes, while others carried relatively few. Notably, isolate_1 exhibited the most extensive resistome profile among all samples.
Associations between STs and plasmid/resistance elementsTo explore potential associations between STs and plasmid/resistance elements, Fisher’s exact test was performed. p-values are reported to highlight possible associations in this study because the multiple-testing correction was overly conservative and adjusted p-values were generally not significant due to tthe large number of comparisons. A number of plasmid elements were found to be distributed among ST17 isolates, including rep1_1_repE(pAMbeta) (p = 0.0015), repUS57_1_rep(AUS0004p2) (p = 0.0020), and repUS7_1_rep(pHTbeta) (p = 0.0087). Additional association included rep14b_4_EMQU3221(pQY003), VanHAX, aph(3’)-III, and β-lactamase genes (blaSHV-107, blaSHV-11, blaSHV-159) with p-values ranging from 0.023 to 0.047. The ST18 isolates exhibited association with a number of resistance genes and plasmid replicons, including aac(6’)-Ii, cat(pC233), lnu(B), and lsa(E) (p = 0.023–0.047), VanHAX, aph(3’)-III, and repUS15_2_repA(pNB2354p1) (p = 0.050). For ST80, significant elements included rep1_1_repE(pAMbeta) (p = 0.033), repUS43_1_CDS12738(DOp1) (p = 0.039), repUS15_1_repA(DO3) (p = 0.033), VanHAX (p = 0.031), aph(3’)-III (p = 0.031), erm(T) (p = 0.017), and tet(S) (p = 0.043). All P-values for the associated elements are presented in Table 1.
Table 1 Association analysis of sequence types (STs) and mobile genetic elements/resistance genesDetection of mobile genetic elements (MGEs)IntegronFinder analysis detected integrons in 13 out of 63 E. faecium isolates examined. The integrase gene (intI), a hallmark of class 1 integrons, was detected in all 13 isolates. Among these, three isolates (Isolate_6, Isolate_16, and Isolate_19) exhibited CALIN (Cluster of attC sites lacking integrase) structures, characterized by the presence of multiple attC recombination sites and associated coding sequences in the absence of a nearby intI gene on the same contig. Isolate_6 showed a CALIN structure consisting of two attC sites and a downstream coding sequence, along with an intI gene present on a separate contig. Isolate_16 displayed a complex CALIN with three attC sites interspersed among multiple ORFs. Isolate_19 carried both an intI gene and a CALIN structure spread across different contigs, suggesting potential modular acquisition or assembly. In the remaining 10 isolates (isolate_1, 2, 3, 4, 5, 7, 9, 11, 12, 18) intI genes were detected without any adjacent attC sites.
Genome-wide detection of IS was performed using ISEScan, identifying a total of 25 distinct IS families across all isolates. The most abundant family was IS3, with 1,303 copies, followed closely by ISL3 (1,250), IS30 (968), and IS256 (944). Other commonly represented families included IS6 (612), IS200/IS605 (422), and IS66 (336), while less abundant families such as IS1 (38), ISA51 (29), and ISKRA4 (15) were comparatively rare. In addition, several unclassified IS elements labeled as “new” were detected (Fig. 7a, S3_Supplementary File), further highlighting the diversity of mobile genetic elements in the studied E. faecium genomes.
To assess the potential mobility of integron-associated regions, the positions of IS elements were cross-referenced with integrons. The IS elements located on the same contig and within ± 10 kb of integrons were considered potentially associated. Notably, in Isolate_1, an IS6-family element (position 560–1379) overlapped with an integron (position 3–578) on contig Isolate_1_169. In Isolate_9, an IS91-family element was located upstream (59–1495) of the integron (1445–1978) on contig Isolate_9_153, falling within the defined 10 kb window. Similarly, in Isolate_12, an IS6-family element (83–902) was found just upstream of the integron (910–1383) on the same contig. These observations suggest potential horizontal gene transfer mediated by IS elements in proximity to integrons, highlighting their possible role in the dissemination of resistance determinants among these isolates.
The IS element profile of E. faecium collection was dominated by IS families, which are known to contribute to genome plasticity, especially in clinical settings. Notably, several antimicrobial resistance genes were often co-localized with IS elements on the same contigs, which support their potential role in resistance gene dissemination. The vanHAX operon, conferring resistance to vancomycin and teicoplanin, was commonly found adjacent to IS families including IS3, ISL3, IS30, IS6, IS256, and IS1380. Aminoglycoside resistance genes, such as aac(6’)-Ii and aac(6’)-aph(2’’), were often located near IS200/IS605, IS66, IS6, and IS256 elements. Similarly, macrolide resistance gene msr(C) and beta-lactam resistance gene blaCTX-M-15 were found in proximity to IS elements, particularly those belonging to the IS3 and IS200/IS605 families. The co-localization of IS elements with resistance genes suggests potential mobility of these loci, which could be explored further to understand gene transfer dynamics in E. Faecium. Complementing this, transposons such as Tn602, Tn6261, Tn6292, Tn4001, Tn5801-like, and Tn5403 were also identified, among which Tn917 was found in the majority of the isolates, further emphasizing the active genomic remodeling events occurring in these strains. These mobile genetic elements likely facilitate horizontal gene transfer and contribute to the dissemination of resistance traits. In contrast, Tn1546 which is a transposon commonly linked with vancomycin resistance, was found in only four isolates (1, 38, 41, and 50). Collectively, the widespread presence of both IS elements and transposons, and their close association with resistance genes, underscores the dynamic nature of the E. faecium genome and its adaptability in hospital-associated environments.
Identification and classification of prophagesThe phage region was estimated using PHASTER, which revealed different prophage content in 63 clinical isolates. PHASTER analysis identified several phage regions with different completeness. The incomplete prophage region was the most abundant, accounting for approximately 36.8% of the total, followed by the questionable region at 32.4% and the intact region at 30.9 (Fig. 7b). This data suggests that a large proportion of prophages may be present in the isolates. In addition, several types of prophages were identified. Of these, the most frequently detected was PHAGE_Lister_2389, which was found in 41 instances in different isolates. This was followed by PHAGE_Staphy_SPbeta_like (33 instances) and PHAGE_Entero_phiFL1A (9 instances). Other phages, including PHAGE_Bacill_BCJA1c and PHAGE_Entero_IME_EFm5, were also commonly observed (Fig. 7c). This study suggests a high degree of prophage diversity in Enterococcus isolates and suggests the for horizontal gene transfer events mediated by phages. Furthermore, phage-inducible chromosomal islands (PICIs) were identified in several isolates based on the presence of integrase, att site specificity, small genome size (< 30 kb), and absence of structural phage genes such as capsid or portal proteins. These regions are known mobile genetic elements that are capable of hijacking helper phages for transfer and may play a role in virulence or antibiotic resistance dissemination. A table with PHASTER results is provided in the S4_Supplementary File.
Co-localization network of plasmid replicons, resistance genes and IS elementsA network analysis was performed utilizing contig-based co-localization data to determine the associations among AMR genes, plasmid replicons, and IS elements in E. faecium isolates. In the network, nodes represent AMR genes, IS elements, or plasmid replicons, whereas edges represent their co-localization on the same contig (S2_Supplementary File [Figure S5]). To identify nodes potentially central to co-localization patterns, five centrality metrics such as Betweenness, Bottleneck, Closeness, Degree, and MCC were calculated, and the top 10 nodes for each metric were analyzed (Table 2 and Fig. 8 (a-e)). The analysis revealed that IS elements including IS1380, IS3, IS200/IS605, IS6, and ISL3 consistently ranked among the top 10 nodes across multiple metrics. Plasmid replicons, such as rep22_1b_repB(pAMalpha1), repUS43_1_CDS12738(DOp1), repUS15_2_repA(pNB2354p1), and rep2_1_orf1(pRE25), were also highly central in Closeness and MCC measures. Among AMR genes, aac(6’)-Ii, aac(6’)-aph(2’’), erm(B), msr(C), and blaTEM-1B were observed in the top 10 nodes for Betweenness, Degree, or MCC. These results indicate a network structure in which certain IS elements, plasmids, and AMR genes are repeatedly co-localized, forming highly connected nodes. These observations provide a framework for additional research on horizontal gene transfer trends in E. faecium and enable the development of testable hypotheses about potential co-localization patterns, such as which IS elements and plasmids are commonly associated with particular resistance genes.
Table 2 Top-ranking nodes in the co-localization network of AMR genes, plasmid replicons, and IS elements in 63 E. faecium isolatesIdentification and functional prediction of resistance-associated mutationsIn addition to acquired resistance genes, this study identified point mutations associated with AMR using the PointFinder tool. The most frequently observed mutations were located in the gyrA and parC genes, followed by pbp5. All isolates exhibited chromosomal mutations in gyrA, parC, and pbp5. Phenotypic resistance to nalidixic acid and ciprofloxacin corresponded with mutations in gyrA and parC, while ampicillin resistance was associated with mutations in pbp5 across all isolates. Due to limitations of PointFinder in identifying specific mutations, we further utilized SnpEff to identify additional mutants, particularly those associated with daptomycin resistance.
Previous studies have shown that daptomycin resistance in Enterococcus faecium is often associated with mutations in specific chromosomal genes, including liaFSR (encoding a three-component regulatory system), eatA (encoding an ABC-F family protein), rpoB and rpoC (encoding the β and β′ subunits of DNA-directed RNA polymerase), and cls (encoding cardiolipin synthase)(Munita et al. 2012; Heidary et al. 2018). In the present study, SnpEff analysis identified multiple mutations in key regulatory and resistance-associated genes. Notably, co-occurring, previously reported resistance-associated amino acid substitutions were detected, including W73C (c.219G > T) in liaR (chromosome position 853742) and T120A (c.358 A > G) in liaS (chromosome position 852807). Additional mutations included T298S (c.892 A > T) in cls (chromosome position 854419), T450I (c.1349 C > T) in eatA (chromosome position 604333), H486Y (c.1456 C > T) and S491F (c.1472_1473delCAinsTC) in rpoB at chromosome positions 2,747,014 and 2,746,997, respectively.
In addition to these literature-reported substitutions, several novel mutations were identified in vancomycin-resistant isolates (Table 3). To assess their potential functional impact, PROVEAN analysis was performed for all amino acid changes. Of these, three mutations: R424S (c.1270 C > A) in cls (chromosome position 854041), M475V (c.1423 A > G) in rpoB (chromosome position 2747047), and T634K (c.1901 C > A) in rpoC (chromosome position 2742873) were predicted to be deleterious (PROVEAN score ≤ − 2.5). Both the previously reported resistance-associated mutations and the PROVEAN-predicted deleterious variants are summarized in Table 3, as they are more likely to impair protein function and contribute to antimicrobial resistance. The distribution of mutations across individual isolates is depicted in Fig. 9 as a bubble heatmap.
Formal statistical analysis was performed to evaluate the relationship between these mutations and the presence of vancomycin resistance genes in 63 isolates. Fisher’s exact test was used to formally analyze the association between these mutations and the existence of the van gene, while multiple comparisons were then taken into account using the Benjamini-Hochberg FDR correction. The Cls-R424S mutation demonstrated a marginal negative association with vancomycin resistance (p = 0.019, adjusted p = 0.057). In contrast, RpoB-M475V, which was found exclusively in vancomycin-resistant isolates and showed an infinite odds ratio, did not reach significance (adjusted p = 0.31). Similarly, RpoC-T634K was frequently found in association with the van gene (OR = 2.49, adjusted p = 0.25), but was not statistically significant (Table 4). These findings indicate that while some missense variants co-occur with resistance determinants, vancomycin resistance in this group likely arises from a multifactorial genomic background rather than from these individual mutations.
Further analysis using MutPred2 was performed to predict the potential molecular mechanisms and functional consequences associated with the identified missense mutations. A mutation was considered deleterious if the MutPred2 score was ≥ 0.5 (Al-Kindi et al. 2020). Except for the M475V variant in RpoB, all other mutations were predicted to be associated with various abnormal molecular mechanisms, suggesting their potential role in resistance. Additionally, DynaMut2 analysis indicated that these missense single nucleotide polymorphisms (SNPs) could destabilize the corresponding proteins, as evidenced by negative ΔΔG values. The predicted molecular mechanisms, associated P-values, and changes in protein stability are summarized in Table 5. The destabilizing effects were further supported by altered intermolecular interactions in the mutant structures compared to their wild-type counterparts (Joshi et al. 2024). In the Cls-R424S mutant, a notable loss of ionic, hydrophobic, and hydrogen bonding interactions was observed relative to the wild-type protein structure. For the RpoB-M475V mutation, a gain in hydrogen bonding and a concurrent loss of van der Waals interactions were detected. Similarly, in the RpoC-T634K mutant, the loss of a hydrogen bond and the gain of a polar interaction were observed, interactions that were not present in the wild-type structure. These structural alterations are illustrated in the Fig. 10.
Table 3 Literature-reported and PROVEAN-predicted deleterious mutations identified in antimicrobial resistance-associated genesTable 4 Association between specific mutations and Vancomycin resistance in Enterococcus faecium isolatesTable 5 Summary of predicted molecular mechanisms, associated P-values, and changes in protein stability for identified novel mutations
Comments (0)