
Remember me

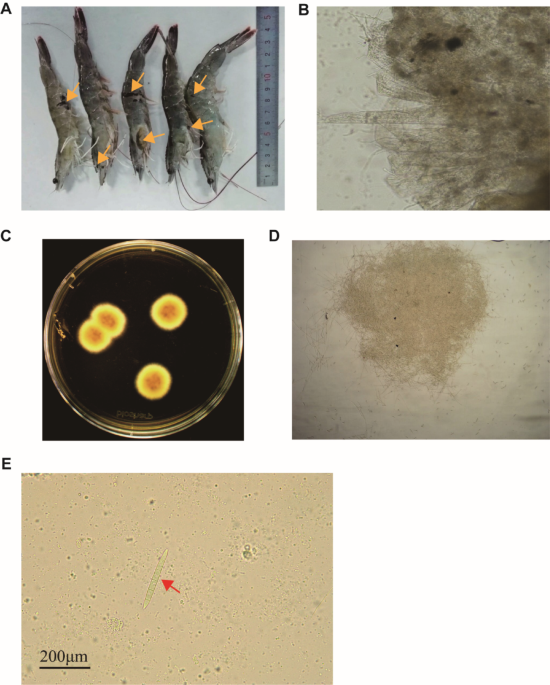

To investigate the genome of 1-Jan, we initially isolated the Fusarium from infected prawns, which exhibited black plaques throughout the body, including the head, chest carapace, abdomen carapace edge, swimming foot, and tail fan edge (Fig. 1A). These black plaques varied in size, with some being perforated. Microscopic examination revealed that parts of the gills and the inner surface of the carapace were filled with hyphae and crescent-shaped spores (Fig. 1B). The isolated fungi could form colonies on PDA plates (Fig. 1C), and the colonies were composed of hyphae and spores (Fig. 1D), the latter of which contained three visible separations (Fig. 1E).
To investigate the causal relationship between infection with the 1-Jan strain and prawn pathogenesis, we challenged healthy P. vannamei with graded concentrations of fungal spores and monitored the clinical symptoms. After an 8-day observation period, infected shrimp developed black gill disease, mirroring the symptoms observed in naturally diseased individuals (Fig. S1A, B). Microscopic examination of gill filaments revealed abundant fungal hyphae in the infected group (arrows in Fig. S1D), while no abnormalities were detected in the control group (Fig. S1C). Furthermore, the infection significantly compromised shrimp survival, indicating a severe pathogenic effect (Fig. S1E). Molecular identification of the fungal strain re-isolated from the infected shrimp confirmed it was genetically identical to the original challenge strain. Collectively, these results demonstrate that the 1-Jan fungal strain is the causative agent of the observed disease symptoms in P. vannamei.
Next, we performed whole-genome sequencing on the 1-Jan strain. The original data underwent a series of filtration steps to generate optimal subreads (Fig. 2A and B; Table 1). In total, we generated 873,391 subreads, corresponding to 9,825,872,319 bp of data. These subreads were assembled using SOAPdenovo software into 54 contigs, with a total length of 51,833,033 bp. To confirm the quality of sequencing and processing, the subreads were back-aligned to the assembled genome. We statistically analyzed the GC content of the sequences (51.73%) and the read coverage depth (62x, Fig. 2C). Detailed statistics of the assembled genome are shown in Table 1.
Fig. 1 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Isolation and characterization of the 1-Jan strain from Fannabin prawns. A Fannabin prawns infected with the 1-Jan strain exhibit black plaques (indicated by orange arrows) throughout the body. B Gill and inner surface of the carapace filled with hyphae. C Fungus colonies on PDA plates. D Hyphae observed within the colonies. E Image showing a spore (red arrow)
Fig. 2 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Characteristics of sequencing assemblies of the 1-Jan strain. Nucleotide content (A) and quality of the sample (B). C The sample GC content and depth analysis chart. The horizontal axis indicates the GC content, and the vertical axis indicates the sequencing depth. The right panel shows the sequencing depth distribution, and the upper panel shows the GC content distribution. D Gene length distribution chart
Table 1 The characteristics of assembly scaffolds and genome of the 1-Jan strainCoding gene predictionTo identify the coding genes in the 1-Jan genome, we analyzed the assembled genome using the Augustus software (Fig. 2D), and the results are summarized in Table 1. The 1-Jan genome contains 9,769 genes with an average length of 1,282 bp, accounting for 24.17% of the whole genome. The predicted coding genes were then analyzed with the NR, Gene Ontology (GO), KEGG, KOG, and Swiss-Prot databases to predict their functions. Specifically, NR provides a comprehensive analysis, resulting in the annotation of 94% of the predicted proteins (Table 2, Supplemental file 1 and 2). GO analysis revealed that 58.8% of the predicted proteins have corresponding GO annotations (Supplemental file 1 and 3), which could be divided into three categories: (1) “Cellular component” describes subcellular structures, locations, and macromolecular complexes, with “membrane part,” “membrane,” and “cell” containing the most annotations; (2) “Molecular function” describes the function of a gene or gene product, with “catalytic activity” and “binding” corresponding to most genes; and (3) “Biological process” describes the processes involving the product encoded by a gene, with “cellular process,” “metabolic process,” and “localization” having the most abundant predicted proteins (Fig. 3).
Fig. 3 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Categorization of all the genes of the 1-Jan strain. The genes are classified based on the GO categories including cellular component, molecular function, and biological process
Table 2 Number of predicted proteins of the 1-Jan strain genome and annotation numbers in respective databasesThe results of KEGG analysis uncovered that 43.9% of the predicted proteins within the 1-Jan genome correspond to KEGG annotations (Table 2, Supplemental file 1 and 4). The most significant annotations were “Transport and catabolism” in “Cellular processes”, “Signal transduction” in “Environmental information processing”, “Translation” in “Genetic information processing”, “Amino acid metabolism” and “Carbohydrate metabolism” in “Metabolism”, and “Infectious diseases” in “Human diseases” (Fig. 4).
Fig. 4 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.KEGG classification of predicted proteins in the 1-Jan strain. Distribution of annotated proteins from the 1-Jan genome according to functional classes in the KEGG database
The KOG database, an orthologous database of eukaryotes, is a protein database created and maintained by NCBI based on the evolutionary relationships of the encoded proteins of the complete genomes of bacteria, algae, and eukaryotes [31]. By aligning, a specific protein sequence can be annotated into a cluster of orthologous proteins, which is composed of orthologous sequences to infer the function of the queried sequence. The data analyzed by KOG indicated that 22.0% of the predicted proteins of the 1-Jan genome could be aligned to a cluster (Table 2, Supplemental file 1 and 5). The functional clusters enriched with the most genes were “Amino acid transport and metabolism”, “Energy production and conversion”, “Translation, ribosomal structure, and biogenesis”, and “Posttranslational modification, protein turnover, chaperones” (Fig. 5).
Fig. 5 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.KOG classification of proteins in the 1-Jan strain. Distribution of annotated proteins from the 1-Jan genome according to functional classes in the KOG database
Swiss-Prot is a database of selected protein sequences that provides a high level of annotation, including a description of protein function, its domain structure, post-translational modifications, and variants, with a minimum level of redundancy compared to other databases and a high level of integration [32]. Swiss-Prot analysis illustrated that 32.9% of predicted proteins could be mapped in the Swiss-Prot (Table 2, Supplemental file 1 and 6). These results collectively demonstrate that the 1-Jan strain genome encodes a significant number of coding proteins that may regulate various biological processes.
Repeat sequence analysisTo explore the presence of repeated sequences in the 1-Jan genome, we performed bioinformatic analysis and the results showed that the 1-Jan genome contains 699 long interspersed nuclear elements (LINEs) and 14 short interspersed nuclear elements (SINEs). In addition, 1,503 long terminal repeats (LTRs), 1,570 DNA transposons, and 67 rolling circles were identified by the genomic analysis (Table 3). Additionally, the 1-Jan genome was predicted to contain 5,199 tandem repeats (TRs), along with 3,987 minisatellites and 471 microsatellites (Table 4).
Table 3 Number of repeat sequences predicted of the 1-Jan strain genomeTable 4 Number of predicted tandem repeats (TRs), minisatellite dnas, and microsatellite DNAs in the 1-Jan strain genomeNon-coding RNA analysisWe then analyzed the distribution of non-coding RNAs (ncRNAs) within the 1-Jan genome. For fungi, ncRNAs mainly refer to small RNA (sRNA), small nucleolar RNA (snoRNA), microRNA (miRNA), transfer RNA (tRNA), and ribosomal RNA (rRNA), with tRNA and rRNA being the most common. Our analysis results showed that the 1-Jan genome has 306 tRNAs, 67 5 S rRNAs, 22 18 S rRNAs, 20 28 S rRNAs, 2 sRNAs, and 10 snRNAs (Table 5).
Table 5 Number of predicted non-coding RNAs in the 1-Jan strain genomePseudogene analysisA pseudogene is a sequence that is similar to a functional gene but contains many mutations. The assembled 1-Jan genome was compared to the protein sequences of the reference species, which were selected based on the comparative genomic studies discussed later. The results showed that all three reference genomes (N. haematococca, F. solani IMV00293, and F. solani JS-169) contained a large number of pseudogenes compared to the 1-Jan genome, suggesting a less conserved evolution of Fusarium fungi (Table 6).
Table 6 Number of predicted pseudogenes of the 1-Jan strain genomeFungal characteristics analysisThe fundamental differences in the genome reflect the distinct functions of various fungi. Using multiple methods, we predicted and annotated relevant databases concerning pathogenic genes and drug resistance, obtaining the specific composition of the 1-Jan genome (Table 7).
Table 7 Number of annotations in various databases describing fungal characteristicsPathogen and host interaction (PHI) database annotationThe content of PHI is experimentally verified, primarily from fungal, oomycete, and bacterial pathogens [33]. Infected hosts include animals, plants, fungi, and insects. The database also includes antifungal compounds and corresponding target genes. Each gene in the database contains nucleic acid and amino acid sequences, as well as a detailed description of the predicted protein function during host infection. The 1-Jan genome generates 1,101 annotations from the database (Table 7, Supplemental file 7), with relevant categories including “effector_(plant_avirulence_determinant),” “enhanced_antagonism,” “increased_virulence_(hypervirulence),” and “lethal” (Fig. 6). The most significant alignment was GFA1 in the “lethal” category, a gene expressed in Aspergillus fumigatus, a prevalent airborne filamentous fungal pathogen in humans, causing severe invasive and even lethal infections in immunocompromised patients [34].
Fig. 6 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.PHI classification of proteins in the 1-Jan strain. Distribution of annotated proteins from the 1-Jan strain genome according to functional classes in the PHI database
Fungal cytochrome P450 database (FCPD) annotationThe FCPD is a comprehensive resource for the P450 family of oxidases, which are superfamily enzymes related to the production and modification of various compounds [35]. A total of 126 predicted proteins in the 1-Jan genome are annotated in the FCPD database (Table 7, Supplemental file 8), with CYP577A1 from Magnaporthe grisea generating the second most significant alignment.
Fungal virulence factor database (DFVF) annotationDFVF is an extensive online resource of known fungal virulence factors, collecting 2,058 disease-causing genes from 228 fungal strains across 85 genera [36]. According to DFVF, the 1-Jan genome produces 479 annotations in the DFVF database (Table 7, Supplemental file 9), with the GFA1 gene of Sporothrix schenckii, being the most significantly aligned.
Carbohydrate-Active enzymes (CAZy) database annotationCAZy database is a specialized resource for carbohydrate enzymes that catalyze the degradation, modification, and biosynthesis of carbohydrates [37]. The predicted proteins from the 1-Jan genome generate 497 CAZy annotations (Table 8), including 225 glycoside hydrolases (GHs), 72 glycosyl transferases (GTs), 35 polysaccharide lyases (PLs), 18 carbohydrate esterases (CEs), and 75 auxiliary activities (AAs). In addition to catalytic domains, CAZy also contains functional domains such as the Carbohydrate-Binding Module (CBM). The 1-Jan genome encodes 72 CBM domains (Table 8, Supplemental file 10).
Table 8 Number of annotations in the CAZy databaseSecreted protein annotationMany secreted proteins are enzymes essential for fungal lifestyle. The N-terminus of secreted proteins includes a signal peptide consisting of 15 to 30 amino acids [38]. Using the signal peptide prediction tool SignalP, 538 proteins encoded by the 1-Jan genome were identified as secreted proteins (Table 7, Supplemental file 11).
Comparative genomic SNP and phylogenetic analysisTo identify SNPs specific to the 1-Jan strain, we compared the 1-Jan genome with eight reference genomes including F. solani JS-169, F. solani IMV00293, N. haematococca, F. virguliforme, F. graminearum, F. verticillioides, F. oxysporum, and F. proliferatum. Subsequently, the alignments were sequentially examined by BLAST, RepeatMasker, and TRF software to obtain reliable SNPs, which are listed in Table 9 and Supplemental file 12.
Next, we used the SNP matrix to construct a phylogenetic tree (Fig. 7), which indicates the evolutionary relationship between species sharing a common ancestor. The phylogenetic tree also shows that the 1-Jan strain is in the same branch as F. solani JS-169, N. haematococca, and F. virguliforme (Fig. 7).
Fig. 7 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.The phylogenetic tree of Fusarium species. The number on the branch indicates the reliability of the branch, with values closer to 100 indicating higher reliability. The length of the branch indicates the evolutionary distance, calculated by the average number of times each nucleotide was replaced. The tips of the tree represent species, and the intersections represent common ancestors between species
Table 9 The SNPs in the genomes of 1-Jan and 8 reference strainsCollinearity analysisThe evolutionary distance between species can also be measured by the degree of collinearity, referring to the genetic linkage relationship, where homologous genes on chromosomes of different species are arranged in the same order. The collinearity between the 1-Jan genome and the eight Fusarium reference genomes was compared and shown in Fig. 8. This indicated that the target strain 1-Jan and the reference strain N. haematococca have the highest collinearity and similarity, with F. solani JS-169 being the second highest. The results here are consistent with the phylogenetic tree.
Fig. 8 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Genome-wide collinearity results showing whole-genome two-dimensional collinearity. The horizontal axis represents the 1-Jan genome, and the vertical axis represents the reference genome. Red lines indicate positive alignment and blue lines indicate reverse alignment. The colors indicate alignment types: Collinearity (collinear alignment), Translocation (translocation alignment), Inversion (inverted alignment), and Tran + Inver (translocation and inverted alignment)
Orthological gene family annotationTo assess the evolutionary relationship between 1-Jan and other Fusarium species, we performed an orthologous analysis. Predicted proteins from 1-Jan were compared in a pairwise fashion to those of eight Fusarium species with available genomic data, including N. haematococca, F. solani IMV00293, F. solani JS-169, F. oxysporum, F. proliferatum, F. verticillioides, F. graminearum, and F. virguliforme using BLAST software. Redundant sequences were removed using Solar software, and Hcluster-sg was subsequently used to cluster proteins based on sequence similarity. We generated a total of 12,699 gene families, of which 3052 were single-copy gene families (one gene copy in each genome, Table 10, Supplemental file 13). The analysis also showed that most of the genes in the studied fungi are orthologs, which are a group of genes originating from the same ancestor and performing the same function in different species. Around half of the orthologous genes were single-copy genes, while the other half were multi-copy genes. The latter are generated during evolution when the genomic sequences are duplicated. Some of these duplications continue to evolve and become new genes different from the original gene, while others remain in the same structure and function. The fungal genomes also contain a small number of paralogs, which are genes duplicated by the same gene in the same species (Fig. 9; Table 10).
Fig. 9The number of homologous genes. Single-copy orthologs: number of single-copy homologous genes in a species-shared gene family; Multiple-copy orthologs: number of multi-copy homologous genes in a species-shared gene family; Unique paralogs: genes in the species-specific gene family; Other orthologs: all other genes; Unclustered genes: genes not clustered into any family
Table 10 Orthological gene family analysisSecondary metabolite (SM) gene cluster analysisSMs are bioactive small molecules not essential for organism growth but important for fungal lifestyles, such as antibiotics produced by biocontrol fungi and phytotoxins synthesized by plant pathogens [39, 40]. The genes required for SM synthesis are usually arranged in a multigene biosynthetic gene cluster in fungi [41]. The predicted results of known secondary metabolic gene clusters showed that the type I polyketide synthases (PKSs) gene cluster Fusarubin is shared among the four strains, while Gibepyrone A is not in 1-Jan, but in the other three strains, as indicated by the antiSMASH analysis data (Table 11 and Supplemental file 14).
Table 11 Secondary metabolic gene clustersIndel detection and annotationIndel is a general term for the insertion and deletion of DNA sequences, and the general indel represents a small indel of 1 to 50 bp. In the genomic coding region, the occurrence of indel may cause frameshift mutations, amino acid substitutions, and the appearance of pseudogenes (Supplemental file 15). We used LASTZ software and a series of filtration strategies to compare the genomes of F. solani IMV00293, F. solani JS-169, and N. haematococca with the 1-Jan genome as the reference. The indel results are summarized in Table 12.
Table 12 InDel frequencies in F.solani.IMV00293, F.solani.JS-169, and N.haematococca with the 1-Jan strain as referencePathogenic factors analysisFinally, we analyzed the potential pathogenic factors by combining the pathogenic factor annotation (Table 7) with the comparative genomic analysis. There are 737 genes in the 1-Jan genome with SNP mutation sites, of which 241 are potential pathogenic factors (Supplemental file 12). The indel analysis of F. solani IMV00293, F. solani JS-169, and N. haematococca with the 1-Jan strain as the reference sequence found 3237 genes in F. solani JS-169 have indel sites, 736 of which are pathogenic factors; 74 genes in F. solani IMV00293 have indel sites, 38 of which are pathogenic factors; 7447 genes in N. haematococca have indel sites, 1637 of which are pathogenic factors (Supplemental file 15).
Since fungal genomes contain multiple orthologs and paralogs (Table 10), we classified genes into clusters, which represent groups of genes with greater than 40% similarity and a sequence length difference of less than 0.4. By comparing the gene clusters in the four genomes of 1-Jan, N. haematococca, F. solani IMV00293, and F. solani JS-169, we defined core gene clusters as the consensus genes in all four samples (Fig. 10 and 501 core gene clusters). After removing the consensus clusters, we obtained the dispensable clusters, and the specific gene clusters are those uniquely possessed only by one genome. All dispensable clusters combined with consensus ones were defined as pan-gene clusters (21,539 in total). Among them, the core and specific gene clusters correspond to the commonality and characteristics of the genome, respectively. Of the core gene clusters, 4,255 single-copy ones were found (single-copy in each genome), 859 of which are potential pathogenic factors (Supplemental file 16).
Fig. 10 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Venn diagram of the genes in four genomes (1-Jan, N. haematococca, F. solani IMV00293, and F. solani JS-169). Each ellipse represents a genome, and the numbers in each region represent the number of gene clusters that appear only in that region. A cluster represents a group of genes with greater than 40% similarity and a sequence length difference of less than 0.4
Besides, there are 1208 specific gene clusters in the 1-Jan genome, corresponding to 1293 genes. These were further filtered according to species annotation information, resulting in 967 specific genes in the 1-Jan genome (Supplemental file 17). Combined with pathogenic factor annotations, 95 unique pathogenic genes in the 1-Jan strain were identified. The 95 genes were classified by KEGG analysis, with 39 genes annotated to known functions (Supplemental file 17). Interestingly, gene 1-JanA2947 (KFY15158.1 in NR) was annotated as a chitinase in the KEGG database, corresponding to the perforated black plaques observed in the prawn body. Further analysis revealed that all the unique pathogenic genes in the 1-Jan genome with CAZy annotation are related to chitin degradation (Supplemental file 18), suggesting that the chitin degradation process might play a critical role in the pathogenesis of the 1-Jan strain in prawns. Figure 11 shows the location of all genes in the 1-Jan genome, GC content, and the location of the pathogenic genes in the genome. Together, these findings identify potential pathogenic genes within the 1-Jan genome and underscore the role of chitinase-related mechanisms in the pathogenesis of this Fusarium species.
Fig. 11 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.The location of GC content, coding genes, and pathogenic genes in the 1-Jan genome. The first section is the GC content of the sequence; the second section shows the distribution of all genes, with blue dots indicating genes on the positive strand and yellow dots indicating genes on the negative strand; the third section represents all pathogenic genes; and the fourth section indicates the positional information on the sequence
Comments (0)