Intrapulmonary delivery of rSIV.F/HN has been previously characterised, with promising features including persistent pulmonary gene expression (at least 2 years) following a single dose, efficient and highly localised transduction of the respiratory epithelium and feasible repeat administration at daily and monthly intervals without loss of transduction efficacy [5,6,7]. As a result, administration of rSIV.F/HN has progressed into a first-in-human trial in CF subjects. Importantly, the vector may also hold promise for other disease indications. More recently, rSIV.F/HN delivered topically to murine lungs successfully achieved therapeutically relevant plasma protein levels of the secreted protein Factor VIII [8].
To investigate further the suitability of rSIV.F/HN vector for treatment of systemic diseases, intravenous administration to wildtype mice was investigated and compared with intrapulmonary administration. We show that a single intravenous dose of rSIV.F/HN achieved sustained expression of protein in sera for the duration of the study (12 months). The biodistribution of vector following intravenous infusion was widespread, with significant protein expression measured in the lung, liver and spleen. rSIV.F/HN was repeatedly administered to assess if intravenous vector delivery provokes an anti-vector immune response. A reduction in luciferase expression to background levels was observed in the lung, liver and spleen following three intravenous doses, accompanied by the generation of significant anti-SIV.F/HN NAbs in sera. Together, these data demonstrate that rSIV.F/HN can be administered intravenously to achieve efficient and sustained systemic protein expression (at least 12 months). However, the generation of significant anti-vector NAbs and loss of protein expression show that repeat intravenous administration is not tolerated, and intrapulmonary delivery of rSIV.F/HN should instead be considered if repeat administration is required.
Systemic delivery of lentiviral vectors has historically shown modest efficacy in animal models. Intra-portal delivery of VSV-G FIX lentiviral vectors to dogs with haemophilia B demonstrated long-term reduction in bleeding episodes in treated dogs and up to 1% normal FIX activity [18]. Vector administration was accompanied by acute inflammatory responses and hepatotoxicity that were resolved with prophylactic anti-inflammatory and anti-histamine treatment, an unexpected finding following successful restoration of FIX activity and lack of toxicity in mouse models [18, 19]. Using the same immunosuppression pre-treatment, intravenous administration of VSV-G lentiviral vectors encoding human fumarylacetoacetate hydrolase (FAH) in a pig model of liver metabolic disease, hereditary tyrosinemia type-1 (HT1) led to anaphylactoid reactions associated with elevated innate immune responses, and thus was not well tolerated in pigs [20]. However, when delivered via the portal vein, lentiviral vectors encoding human FAH achieved liver repopulation with FAH-positive hepatocytes in knockout pigs and prevention of precancerous lesions until the end of the study [20]. Although no signs of toxicity were observed in mice that received rSIV.F/HN by intravenous infusion, full characterisation of the innate immune response following vector administration should be performed, particularly in larger animal models, which often do not recapitulate responses in mice. Across the 52-week study, three mice died prematurely in the intrapulmonary group. Two mice were culled based on veterinary advice due to an infected eye wound (7.6e7 TU vector group) and from overgrooming (TSSM group), respectively and one mouse was found dead 36 weeks post-treatment (2.4e7 TU vector group), but the cause was unknown. This mouse was 45 weeks old at the time of death. All deaths were considered incidental rather than treatment-related. This is consistent with our previous study, which demonstrated no evidence of long-term chronic toxicity in mice following intrapulmonary rSIV.F/HN vector administration [6].
In this study, we utilised the human CMV enhancer coupled to the ubiquitous human EF1α short promoter (hCEF), which was previously selected as part of our lead clinical candidate to drive CFTR expression following persistent and efficient gene expression in both the murine nose and lung and human ex vivo air-liquid interface cultures [7]. Following intrapulmonary delivery, it was shown that several cell types within the murine lungs are transduced by the rSIV.F/HN pseudotyped vector, including ciliated epithelial cells, goblet and club cells and type I and II pneumocytes [7]. In human ALI cultures, a similar range of cell types was transduced by rSIV.F/HN, which is not unexpected given Sendai virus envelope protein HN binds to sialylated glycans, specifically α2,3 sialylated N-acetyllactosamine (LacNAc), which is prevalent on human airway cells [21].
The tropism of rSIV.F/HN when delivered systemically should be dictated by the expression of Sendai virus HN receptors, known to be ubiquitous across many cell and tissue types [21]. Successful transduction has been demonstrated with recombinant Sendai vectors in the vascular endothelium, skeletal muscle, cardiomyocytes, upper and lower respiratory tract, brain and retina across multiple species, including rats, mice, ferrets and sheep [22,23,24,25,26,27,28]. The results presented here from the firefly luciferase biodistribution study revealed that systemic delivery of rSIV.F/HN vector results in widespread organ transduction, reflecting previous literature utilising recombinant Sendai vectors. A limitation of intraperitoneal luciferin injection is that not all organs take up the substrate equally, potentially underestimating luciferase expression and vector biodistribution to some organs. To mitigate this limitation, luciferase expression was further quantified in organ homogenate to more accurately assess vector biodistribution. To improve the efficiency of systemically delivered lentiviral vectors and to prevent expression in off-target organs, many have investigated the use of tissue-specific promoters to drive transgene expression, for instance, the use of the alpha-1-antitrypsin (AAT) promoter to drive hepatic expression [20]. Although no specific therapeutic application was investigated here, inclusion of a liver-specific promoter could be assessed in the future to drive hepatic rSIV.F/HN transgene expression for clotting disorders and other similar indications.
Incorporation of an inhibitor of phagocytosis, CD47, in lentiviral vector producer cells was shown to mitigate VSV-G vector clearance by liver phagocytes, reduce recognition by the innate immune system, and enhance hepatocyte gene transfer and FIX activity in non-human primates [29]. Phagocytosis of lentiviral vectors by antigen-presenting cells (APCs) in the liver and spleen therefore, highlights an impediment to systemic vector delivery [30]. Considering significant transduction of these organs is observed following systemic rSIV.F/HN delivery, incorporation of CD47 might be beneficial in the future. Detection of low levels of bioluminescence in the gonads of some mice following intravenous rSIV.F/HN delivery raises an important safety concern and potential impediment to systemic vector delivery due to the risk of lentiviral genome insertion into germ cells and germline transmission. Although bioluminescence was not significantly above background levels in treated mice, it should be established if lentiviral genome can be detected in germ cells following systemic rSIV.F/HN delivery. However, this is unexpected as lentiviral vectors pseudotyped with both Sendai virus F and HN envelope proteins were unable to transduce germline stem cells when cultured ex vivo [31]. Clearly, an advantage of intrapulmonary vector delivery is the containment of vector in the thoracic cavity, shown here by the lack of bioluminescence detected in murine gonads following intrapulmonary rSIV.F/HN delivery.
Vector biodistribution was assessed using lentivirus encoding firefly luciferase and EGFP fusion (rSIV.F/HN-EGFPLux) by luciferase quantification in live mice and organ homogenate and EGFP detection by direct fluorescence microscopy of organ slices to determine cell-type expression. Analysis of liver and spleen tissue slices from mice delivered rSIV.F/HN-EGFPLux by intravenous administration did not reveal any detectable EGFP signal. This is likely due to an overall low transduction efficiency, which makes it difficult to find individual transduced cells in a large organ such as the liver, or quenching of the GFP fluorescence due to fusion with luciferase (data not shown). Cell-type-specific determination of rSIV.F/HN transduction could instead be investigated in the liver and spleen using an EGFP vector and flow cytometry. Alternatively, vector biodistribution could be assessed by RNA extraction from mouse tissue and determination of vector genomes by quantitative PCR. Unfortunately, this was beyond the scope of the current study due to technical limitations. EGFPLux was also selected for repeat administration studies to provide a simple method to compare gene expression between mice that received one and three rSIV.F/HN doses. Control rSIV.F/HN vector encoding mADAMTS13 was selected as an irrelevant vector because we previously concluded that matching doses of rSIV.F/HN mADAMTS13 vector delivered intravenously to mice did not achieve detectable levels of protein (data not shown), and thus was not predicted to influence the phenotype of wildtype mouse.
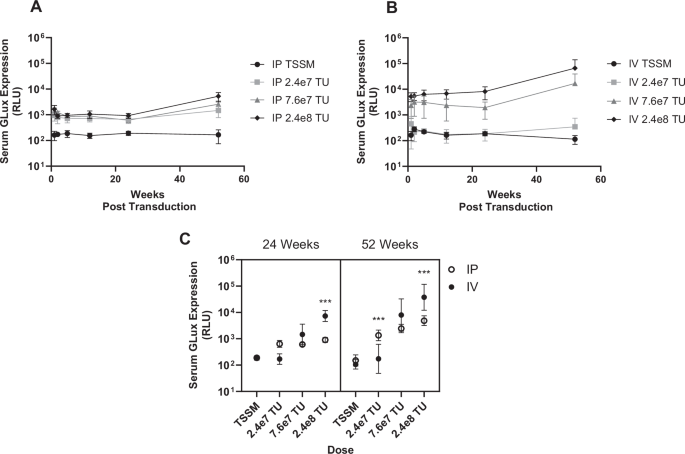
The long-term duration study demonstrated that rSIV.F/HN administered via either the intrapulmonary or intravenous routes leads to stable transgene expression in serum for the duration of the experiment (at least 12 months). This supports previous reports whereby a single intrapulmonary dose of rSIV.F/HN-GLux demonstrated stable expression in sera twelve months post-transduction [8]. The overall trend in both the intravenous and intrapulmonary cohorts was a dose-related increase in transduction efficiency, with dose determined as a significant factor influencing serum expression. The delivery method with superior generation of reporter protein was dependent on the vector dose, as shown by a significant interaction of dose and delivery route. Intrapulmonary delivery was more potent at the lowest dose (2.4e7 TU/mouse), both delivery methods performed equally at the medium dose, and intravenous delivery demonstrated enhanced efficacy (approximately 10-fold) at the highest dose tested (2.4e8 TU/mouse). Vector delivered by intravenous administration demonstrated a threshold dose below which GLux expression was at background levels. This may be due to the vector being diluted among multiple organs following systemic delivery compared with the highly localised transduction of the nose and lungs following intrapulmonary delivery. This study suggests that higher doses of rSIV.F/HN are required to achieve detectable transgene expression in serum following intravenous than intrapulmonary delivery.
Following demonstration of sustained long-term expression following a single dose, rSIV.F/HN was repeatedly administered, whereby luciferase expression in mice that received 3 vector doses was at background levels in the lung, liver and spleen. Here, luciferase expression was quantified at the terminal time-point only, as only the final vector encoded luciferase. However, neutralising antibody titre was monitored longitudinally as sera were sampled from mice after one, two or three vector administrations. Mice that received a single intravenous dose demonstrated a detectable level of neutralising antibodies in sera, and after two doses, a strong induction of neutralising antibodies was observed, which plateaued to the maximum antibody titre following three doses, albeit with limited n numbers. We cannot exclude that cellular immune responses induced after the second and third dose lead to the elimination of transduced cells. However, the experiment was not designed to study cellular immune responses, and instead, we focused on tissue luciferase expression and anti-vector responses with repeat intravenous administration. In contrast, we have shown previously that sustained transduction efficiency is demonstrated following repeat intrapulmonary administration of rSIV.F/HN [6]. One difference in experimental design was the dose delivered to mice (1e7 TU/mouse intrapulmonary, 1.5e8 TU/mouse intravenously), with the 15-fold difference potentially eliciting a stronger immune response due to a greater number of vector particles. It is plausible that a lower dose of vector might be tolerated following repeat intravenous delivery. However, this seems unlikely given that a moderate neutralising antibody response was generated following just one intravenous administration. Rhesus monkeys delivered two intravenous doses of lentiviral vector, noted substantial anti-vector responses after one dose, which completely inactivated the second lentiviral administration [32].
In summary, rSIV.F/HN lentiviral vectors successfully achieved persistent and widespread expression of a reporter protein following a single intravenous dose. Broad tropism may suggest the potential of rSIV.F/HN vector as a therapeutic for wider disease indications. For example, respiratory diseases of the pulmonary endothelium, haematological diseases including clotting disorders, infectious diseases and inherited metabolic disorders, amongst others. The effective transduction of organs and tissues other than the lungs may allow greater flexibility in the selection of transgene, including those which encode proteins that are more efficiently post-translationally modified in organs such as the liver rather than the airways.
The production of a strong anti-vector response following repeat intravenous administration may suggest that systemic rSIV.F/HN delivery is not suited for chronic diseases that require lifelong transgene expression and repeat dosing. However, other therapeutic viral vectors, such as AAV, are equally limited to single administrations due to immune recognition, yet have shown remarkable long-term correction of disease following a single AAV dose [12]. Moving forward, strategies such as tissue-specific promoters could be investigated to enhance transgene expression in desired organs. Alternatively, utilising the lungs as a factory for production of systemic proteins has demonstrated sustained expression of therapeutic transgenes in serum with feasible repeat administration. Intrapulmonary administration might therefore be an efficient yet safer delivery method than systemic delivery moving forward, particularly if the disease application requires long-term gene delivery.

Comments (0)