
Remember me
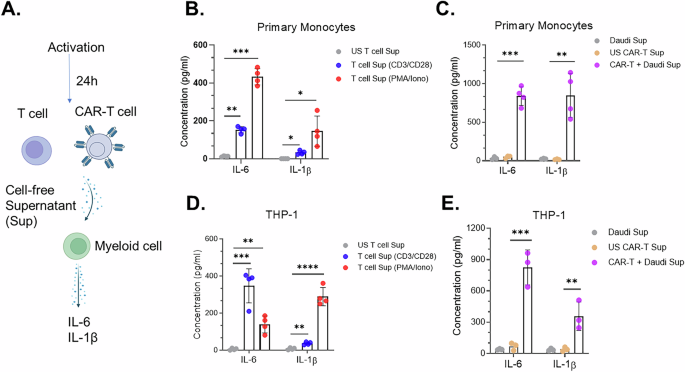
In vitro, chimeric antigen receptor (CAR) T cells can be activated through various methods, including T cell receptor (TCR)-dependent activation with anti-CD3/CD28 antibodies, TCR-independent activation with phorbol myristate acetate (PMA) and ionomycin (Iono), or CAR-mediated activation by co-culturing CAR-T cells with target cells. To determine whether soluble factors secreted by T cells upon activation via TCR-dependent, TCR-independent, or CAR-dependent mechanisms can induce bystander myeloid cell activation (BMCA), primary human T cells or anti-CD19 CAR-T cells were activated using anti-CD3/CD28, PMA/Iono, or co-culture with CD19⁺ Daudi cells. After 24 h, cell-free supernatants were collected and added to myeloid cells, either primary human monocytes or the THP-1 monocytic cell line. BMCA was then assessed after an additional 24 h by measuring IL-6 and IL-1β secretion from the myeloid cells (Fig. 1A).
Fig. 1: Soluble factors secreted by activated T cells and CAR-T cells drive bystander myeloid cell activation. The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.A Schematic of experimental steps followed for myeloid cell activation assay. Primary human T or anti-CD19 CAR-T cells were activated and following 24 h of activation, cell-free supernatant was obtained and incubated with myeloid cells. Myeloid cell activation was measured after 24 h by assessing IL-6 and IL-1β secreted by myeloid cells in the supernatant. IL-6 and IL-1β secreted by primary monocytes following treatment with cell-free supernatants obtained from either unstimulated (US) or activated B T cells and C anti-CD19 CAR-T cells. IL-6 and IL-1β secreted by THP-1 monocytic cell line following treatment with cell-free supernatants obtained from either unstimulated (US) or activated D T cells and E anti-CD19 CAR-T cells. *P < 0.05, **P < 0.01, ***P < 0.001. Each dot represents one donor.
Supernatants obtained from primary T cells activated with anti-CD3/CD28 or PMA/Iono contained high levels of IFN-γ, and GM-CSF compared with supernatants from unstimulated (US) T cells (Fig. S1A). Similarly, supernatants from anti-CD19 CAR-T cells co-cultured with Daudi cells contained high levels of IFN-γ and GM-CSF compared with supernatants from Daudi cells alone or US CAR-T cells (Fig. S1B). These findings confirm robust activation of T cells and CAR-T cells and the presence of IFN-γ and GM-CSF in the collected supernatants.
Next, primary human monocytes were treated with cell-free supernatants obtained from T and CAR-T cells. Supernatants from T cells activated via either TCR-dependent or TCR-independent methods, as well as from activated CAR-T cells, significantly induced IL-6 and IL-1β production in primary monocytes (Fig. 1B). In contrast, supernatants from US T cells, US CAR-T cells, or Daudi cells did not induce cytokine production (Fig. 1C), indicating that myeloid activation is specifically driven by factors secreted by activated, but not resting, T cells and CAR-T cells. Consistent with the data obtained from primary human monocytes, the THP-1 monocytic cell line was also activated by supernatants from activated T cells and CAR-T cells (Fig. 1D, E). To confirm that the IL-6 and IL-1β detected in myeloid and T cell co-culture supernatants originated from myeloid cells rather than from T cells or CAR-T cells, we assessed IL-6 and IL-1β levels in supernatants from unstimulated and CD3/CD28-activated T cells and CAR-T cells. IL-6 and IL-1β were not detectable in supernatants from either unstimulated or activated T cells or CAR-T cells (Fig. S1C). In contrast, IL-6 and IL-1β were detected only in supernatants from activated THP-1 cells and were not detected in unstimulated THP-1 supernatants (Fig. S1C). Collectively, these data indicate that the IL-6 and IL-1β measured in myeloid and T cell co-culture supernatants are derived from activated myeloid cells rather than from T cells or CAR-T cells.
Together, these data demonstrate that upon TCR-dependent, TCR-independent, or CAR-dependent activation, T and CAR-T cells release soluble factors that activate bystander myeloid cells.
Comparative cytokine profiling of CAR-T cells following TCR- and CAR-dependent activation identifies inflammatory candidatesSince supernatants obtained from both TCR-dependent and CAR-dependent activation significantly induced myeloid cell activation (Fig. 1B–E), we hypothesized that shared inflammatory factors may play a central role in BMCA. To identify such candidates, we performed a comparative analysis of cytokines secreted by CAR-T cells following TCR- and CAR-mediated activation.
Anti-CD19 CAR-T cells were generated by activating peripheral blood-derived T cells with anti-CD3/CD28 antibodies, transducing them with an anti-CD19 CAR-expressing lentiviral vector, and expanding them ex vivo. The cell-free supernatant collected at harvest served as the TCR-dependent activation sample. For the CAR-dependent activation sample, anti-CD19 CAR-T cells were co-cultured with CD19⁺ Daudi cells at an effector-to-target (E: T) ratio of 1:1, and supernatant was collected after 24 h. Supernatants from unstimulated (US) CAR-T cells and Daudi cells were used as negative controls.
Prior to cytokine profiling, the supernatants were tested for their ability to induce BMCA. As expected, only supernatants from activated CAR-T cells (both TCR-dependent and CAR-dependent), but not from US CAR-T cells, induced activation of THP-1 myeloid cells (Fig. 2A). We then subjected the supernatants to cytokine analysis. Supernatants from US CAR-T cells or Daudi cells contained minimal levels of inflammatory cytokines (Fig. 2B). In contrast, activation of CAR-T cells resulted in a significant increase in cytokine production, with consistent patterns observed across four independent donors (D1-D4) (Fig. 2B). Although both activation methods generated supernatants that robustly induced myeloid activation with no significant difference in potency (Fig. 2A), total inflammatory cytokine levels were significantly higher in the co-culture supernatant (CAR-dependent activation) than in the harvest supernatant (TCR-dependent activation) (Fig. 2C). We further analyzed 30 inflammatory cytokines individually and found that most were significantly upregulated following CAR-dependent activation compared with TCR-dependent activation (Fig. S2). Together, these data suggest that CAR-dependent activation may induce additional cytokines that do not directly contribute to BMCA.
Fig. 2: Comparative cytokine profiling of CAR-T cells following TCR- and CAR-dependent activation. The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.A IL-6 and IL-1β secretion by the THP-1 monocytic cell line following treatment with cell-free supernatants obtained from either unstimulated (US) or activated anti-CD19 CAR-T cells. Supernatant collected during harvest of anti-CD19 CAR-T cells was used as the TCR-dependent activation sample, while supernatant collected following co-culture of anti-CD19 CAR-T cells with CD19⁺ Daudi cells was used as the CAR-dependent activation sample. B Heatmap of 96 inflammatory cytokines/chemokines present in supernatant samples obtained from CAR-T cells that were either unstimulated, TCR-activated with anti-CD3/CD28 (harvest), or CAR-activated with CD19⁺ Daudi cells (co-culture) at an effector-to-target ratio of 1:1. Cytokine/chemokine levels (expressed as log₁₀ pg/ml) are represented by a color gradient, ranging from blue (no expression) to red (highest concentration). Data from four donors (D1–D4) are shown. C Quantification of 96 cytokines/chemokines detected in the supernatant samples from panel B. Each dot represents a cytokine or chemokine, and the average concentration across four donors is shown. D Quantification of 13 inflammatory cytokines that were absent in the supernatant from US anti-CD19 CAR-T cells but present in the supernatant from TCR-activated (harvest) and CAR-activated (co-culture) anti-CD19 CAR-T cells. *P < 0.05, **P < 0.01, ***P < 0.001. ns= not significant. Each dot represents one donor.
Comparative analysis of cytokines present in the harvest and co-culture supernatants identified 13 cytokines, IFN-γ, GM-CSF, TNFα, IL-13, IL-16, IL-31, MIP-1α, MIP-1β, IP-10, I-309, M-CSF, IL-17A, and IL-17F, that were upregulated ( > 10-fold) relative to unstimulated controls and were common to both TCR- and CAR-dependent activation conditions (Fig. 2D). Because both activation methods induced myeloid cell activation, we focused on these 13 shared cytokines for further analysis, as they likely represent the primary contributors to inflammatory toxicities observed during CAR-T cell therapy.
In addition to these array-identified soluble cytokines, we next evaluated Glycoprotein Ibα (GPIbα) as an additional candidate mediator that was not included in the cytokine profiling panel but was implicated in myeloid activation in our prior CAR-NK studies [33], and we therefore tested whether it could similarly promote bystander myeloid activation following CAR-T cell activation.
T cell-derived GPIbα activates myeloid cellsAs stated above, since our cytokine profiling approach in Fig. 2 was limited to analytes included on the array, we next examined GPIbα, inflammatory mediator not represented in the cytokine array panel but implicated in myeloid cell activation in our prior CAR-NK studies [33], to assess its ability to induce bystander myeloid activation in the current CAR-T cell study.
(GPIbα) is a type I transmembrane protein and the major ligand-binding subunit of the GPIb-V-IX complex, best known for its role as a receptor for von Willebrand factor in hemostasis and thrombosis [34]. Recent studies have shown that soluble GPIbα secreted by activated lymphocytes (T and NK cells) can activate myeloid cells [33, 35].
To assess whether human T cells express and secrete GPIbα, we analyzed both cell lysates and supernatants. Unstimulated (US) T cells expressed GPIbα intracellularly, as it was detected in cell lysates but not in supernatants (Fig. 3A), suggesting no active secretion under resting conditions. In contrast, upon activation, GPIbα was detected in the supernatant, and its intracellular levels were significantly reduced (Fig. 3A, B), indicating that T cell activation triggers GPIbα secretion.
Fig. 3: T cell-derived GPIbα activates myeloid cells. The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.A Immunoblot analysis of cell lysates and supernatants obtained from unstimulated (US) or PMA/ionomycin-stimulated primary human T cells for GPIbα. GAPDH was used as a loading control. B Quantification of cellular GPIbα in lysates from US or PMA/ionomycin-stimulated primary human T cells. Percent (%) cellular GPIbα was normalized to US samples. C Recombinant rGPIbα protein was added to primary monocytes, and IL-6 release in the supernatant was measured by ELISA. Mock-treated primary monocytes received control rGFP protein. D Supernatants obtained from anti-CD3/CD28-activated primary human T cells (CD3⁺, CD4⁺, and CD8⁺) or E supernatants from Daudi co-culture–activated anti-CD19 CAR-T cells were subjected to neutralization using anti-GPIbα antibodies. Following neutralization, supernatants were added to primary monocytes from the same donor, and secretion of IL-6 and IL-1β by myeloid cells was measured. Percent (%) reduction in myeloid cell activation was calculated by normalizing IL-6 and IL-1β levels to those secreted by myeloid cells incubated with IgG control–treated supernatant. F Anti-CD19 CAR-T cell potency was assessed by measuring lysis of CD19⁺ Nalm6 target cells in the presence of control IgG or anti-GPIbα antibodies at various effector-to-target ratios. *P < 0.05, **P < 0.01, ***P < 0.001. Each dot represents one donor.
To determine whether GPIbα directly contributes to myeloid cell activation, we treated primary human monocytes with recombinant GPIbα (rGPIbα). Consistent with prior findings [33], rGPIbα significantly induced IL-6 secretion from monocytes, confirming its inflammatory potential (Fig. 3C).
Next, we investigated whether neutralization of GPIbα secreted by activated T cells could suppress myeloid cell activation. CD3⁺, CD4⁺, and CD8⁺ T cells were isolated from peripheral blood and activated with anti-CD3/CD28 antibodies. Supernatants from these activated T cells were treated with either anti-GPIbα antibodies or control IgG before being applied to primary monocytes. GPIbα neutralization significantly reduced IL-6 and IL-1β secretion from monocytes across all T cell subsets compared with IgG-treated controls (Fig. 3D). These results suggest that GPIbα released by activated CD3⁺, CD4⁺, and CD8⁺ T cells contribute to bystander myeloid cell activation.
We then extended this analysis to CAR-T cells. Anti-CD19 CAR-T cells were activated by co-culture with CD19⁺ Daudi cells, and the resulting supernatants were treated with anti-GPIbα antibodies or control IgG. When applied to primary monocytes, GPIbα-neutralized supernatants led to a significant reduction in IL-6 and IL-1β production compared with IgG-treated controls (Fig. 3E), further confirming the inflammatory role of GPIbα.
Finally, to determine whether GPIbα neutralization impairs CAR-T cell function, we evaluated the cytotoxicity of anti-CD19 CAR-T cells against CD19⁺ Nalm6 cells in the presence of either control IgG or anti-GPIbα antibodies across a range of effector-to-target (E: T) ratios. GPIbα neutralization did not affect CAR-T cell cytotoxicity (Fig. 3F).
Together, these findings demonstrate that GPIbα is released by activated T cells and CAR-T cells, contributes to myeloid cell activation, and that its neutralization reduces inflammatory activation of myeloid cells without compromising CAR-T cell anti-tumor function in an in vitro short-term co-culture assay. Based on these results, we identify GPIbα as a key inflammatory mediator involved in bystander myeloid cell activation.
Neutralization studies confirm the role of CAR-T cell secreted inflammatory factors in myeloid cell activationA total of 14 soluble factors secreted by activated CAR-T cells were identified as potential contributors to myeloid cell activation (Figs. 2 and 3). To assess their functional relevance, we performed antibody-mediated neutralization of each factor in activated CAR-T cell supernatants. Anti-CD19 CAR-T cells from four independent donors were co-cultured with CD19⁺ Daudi cells to generate activated supernatants, and each sample was individually neutralized with factor-specific antibodies. Neutralized supernatants were incubated with myeloid cells for 24 h. IL-6 and IL-1β levels were measured as indicators of myeloid activation, with supernatants treated with non-specific IgG serving as controls. Antibody concentrations were optimized as previously described [33].
Among the 14 factors, neutralization of IFNγ, GM-CSF, GPIbα, or TNFα each led to a ≥ 50% mean reduction in both IL-6 and IL-1β levels (Fig. 4A). In contrast, neutralization of IL-13, IL-16, IL-31, MIP-1α, and I-309 selectively reduced IL-1β but had minimal effect on IL-6. Neutralization of MIP-1β, IP-10, M-CSF, IL-17A, and IL-17F had little to no impact on either cytokine. These findings identify IFNγ, GM-CSF, GPIbα, and TNFα as major inflammatory mediators secreted by CAR-T cells that drive myeloid activation. Our results also confirm prior reports implicating IFNγ, GM-CSF and TNFα as key drivers of CAR-T-associated inflammation [21,22,23,24,25,26].
Fig. 4: Neutralization studies identify the role of four CAR-T cell–secreted inflammatory factors in myeloid cell activation. The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Supernatants obtained from Daudi co-culture–activated anti-CD19 CAR-T cells (effector-to-target ratio of 1:1) were subjected to antibody-mediated neutralization. Following neutralization, the supernatants were added to THP-1 myeloid cells, and secretion of IL-6 and IL-1β was measured. Percent (%) reduction in myeloid cell activation was calculated by normalizing IL-6 and IL-1β levels to those secreted by myeloid cells incubated with IgG control–treated supernatants. Percent reduction in myeloid cell activation following neutralization of A 14 individual soluble inflammatory factors or B combinations of soluble inflammatory factors present in the supernatant from activated anti-CD19 CAR-T cells is shown. Each dot represents one donor.
While each of the four major inflammatory factors, IFNγ, GM-CSF, GPIbα, and TNFα, contributed significantly to myeloid cell activation, neutralization of any single factor was insufficient to fully suppress this response (Fig. 4A). To evaluate potential synergistic effects, we performed combinatorial neutralization studies (Fig. 4B). Dual blockade of IFNγ and GM-CSF, or IFNγ and GPIbα, resulted in comparable reductions in myeloid activation (Fig. 4B), suggesting that GPIbα may contribute to inflammation at a level similar to GM-CSF. In contrast, combined neutralization of GPIbα and TNFα reduced IL-6 production to a similar degree but was notably less effective in suppressing IL-1β (Fig. 4B), indicating that IL-1β induction may rely more heavily on IFNγ and/or GM-CSF.
Addition of a third factor, IFNγ, GM-CSF, and GPIbα, did not further reduce cytokine levels compared to the dual combinations (Fig. 4B), implying that GM-CSF and GPIbα may function through overlapping or convergent pathways. However, simultaneous neutralization of all four factors (IFNγ, GM-CSF, GPIbα, and TNFα) led to a significantly greater reduction in IL-6 levels than any two- or three-factor combination (Fig. 4B), suggesting that comprehensive suppression of IL-6 requires broad inhibition of multiple inflammatory signals.
Paradoxically, this four-factor blockade was less effective in reducing IL-1β levels and mirrored the effect observed with dual neutralization of GPIbα and TNFα (Fig. 4B). These findings indicate that the regulation of IL-6 and IL-1β is governed by distinct cytokine networks, highlighting the complex and multifaceted nature of myeloid cell activation in response to CAR-T cell-derived factors.
Currently, CAR-T cell-associated inflammatory toxicities are primarily managed with corticosteroids or by inhibiting IL-6 and/or IL-1β signaling pathways using tocilizumab (an IL-6 receptor antagonist) or anakinra (an IL-1 receptor antagonist) [4]. In some cases, emapalumab (an anti-IFNγ antibody) or etanercept (a TNFα inhibitor), in combination with these agents, have also been used [19, 20]. Although these immunomodulatory therapies have shown efficacy in reducing CAR-T-related inflammatory toxicities, responses remain heterogeneous, and not all patients benefit [20, 36].
Consistent with this clinical variability, we observed donor-specific differences in the effects of neutralization on CAR-T cell supernatants. For example, in supernatants from donor 1, neutralization of IFN-γ and GM-CSF had minimal impact on IL-6 levels, whereas neutralization of GPIbα and TNFα reduced IL-6 by more than 60% (Fig. S3A). In donor 2, neutralization of IFN-γ and GM-CSF modestly reduced IL-6 (47%) but had minimal impact on IL-1β (8%), while neutralization of GPIbα and TNFα reduced IL-1β by 29% and IL-6 by 87% (Fig. S3B). In both donors, simultaneous neutralization of all four factors substantially reduced both IL-6 and IL-1β (Fig. S3).
Together, these data suggest that due to donor-specific variability, targeting individual inflammatory mediators may not be sufficient to suppress CAR-T-induced myeloid activation. A combinatorial strategy targeting multiple key mediators may therefore be necessary to achieve consistent and meaningful reductions in inflammatory toxicities across a broader patient population.
siRNA-mediated knockdown of major inflammatory factors in CAR-T cells reduces myeloid cell activationThe identification of major inflammatory factors released by activated CAR-T cells that drive myeloid cell activation presents a novel opportunity to enhance safety by genetically modifying CAR-T cells during manufacturing. Although previous studies have identified roles for IFNγ, GM-CSF, and TNFα in mediating inflammatory toxicities during CAR-T cell therapy [22, 23, 26], none have evaluated the effect of simultaneous knockdown of these factors on myeloid cell activation. Furthermore, we demonstrated that GPIbα is a novel soluble factor released by activated CAR-T cells that significantly contributes to myeloid cell activation.
To assess whether knockdown of these four factors, IFN-γ, GM-CSF, TNFα, and GPIbα could reduce myeloid cell activation, anti-CD19 CAR-T cells were transfected with siRNAs targeting these mediators in three different combinations: IFN-γ + GM-CSF, GPIbα + TNFα, and all four factors combined. A non-specific siRNA served as a negative control. After 48 h, CAR-T cells were co-cultured with CD19⁺ Daudi cells. Following 24 h of co-culture, the resulting supernatants were incubated with primary monocytes, and myeloid cell activation was assessed by measuring IL-6 and IL-1β production.
siRNA transfection had no impact on CAR-T cell viability (Fig. 5A). Across four donors, siRNA transfection significantly reduced mRNA expression, with average reductions ranging from 52% to 72% for the targeted genes (Fig. 5B). This was accompanied by significant decreases in protein expression of IFN-γ, GM-CSF, and TNFα, ranging from 63% to 74% (Fig. 5C). GPIbα protein levels were not quantified due to the lack of a suitable assay.
Fig. 5: siRNA-mediated knockdown of four soluble factors in CAR-T cells reduces myeloid cell activation. The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Anti-CD19 CAR-T cells were transfected with the control non-specific siRNA or the pooled siRNAs to reduce GPIbα, GM-CSF, TNFα and IFNγ protein expression. A cellular viability, B mRNA and C protein expression was measured after 48 h of siRNA transfection. siRNA-treated anti-CD19 CAR-T cells were activated following co-culture with Daudi target cells at two different effector to target (E: T) ratios, 1:1 and 5:1. Myeloid (THP-1) cells were incubated with supernatants from siRNA-transfected and activated anti-CD19 CAR-T cells, and IL-6 and IL-1β secreted by myeloid cells were measured. Percent (%) reduction in myeloid cell activation was calculated by normalizing to the D IL-6 and E IL-1β levels secreted by myeloid cells incubated with control siRNA treated and activated anti-CD19 CAR-T cell supernatant. *P < 0.01. ns= not significant. Each dot represents one donor.
In all three combinations, knockdown of these inflammatory mediators in CAR-T cells significantly reduced IL-6 and IL-1β production by monocytes compared to control siRNA treated cells (Fig. 5D, E). No significant differences in cytokine reduction were observed among the three siRNA groups, suggesting that each combination is similarly effective at suppressing myeloid cell activation. While reductions were consistent across the four donors tested, some donor-to-donor variability was observed, as noted in the neutralization studies (Fig. S3). Thus, a combinatorial knockdown strategy targeting all four factors may offer the most broadly effective approach to mitigating CAR-T cell-mediated bystander myeloid cell activation.
It is important to note that the magnitude of the effects observed in our in vitro studies will need to be confirmed in in vivo and other relevant models. Incomplete reduction in myeloid cell activation suggests that these mediators contribute to bystander myeloid activation but are unlikely to function in isolation, consistent with redundancy and compensatory signaling within inflammatory networks. While these data support a contributory role for IFN-γ, GM-CSF, GPIbα, and TNF-α in CAR T-mediated myeloid activation, the modest effect sizes indicate that additional studies are required to achieve more robust mitigation of toxicity. Importantly, our in vitro assays model early, short-term interactions and may underestimate or fail to capture amplifying effects that could occur in vivo or after repeated antigen exposure. Therefore, the translational impact of targeting these factors will require validation in more physiologic models that assess durability, persistence, and systemic inflammatory responses. To evaluate whether gene silencing of inflammatory mediators affects CAR-T cell function, we assessed two key indicators of CAR-T potency: cytokine secretion and cytotoxic activity. Anti-CD19 CAR-T cells transfected with siRNAs targeting IFN-γ, GM-CSF, TNFα, and GPIbα (in the three combinations) were co-cultured with CD19⁺ Daudi cells at two different effector-to-target (E: T) ratios. IL-2 secretion, a marker of CAR-T cell activation and function, was measured 24 h post co-culture. We selected IL-2 instead of IFNγ for potency assessment because measurement of IFNγ in this setting would primarily reflect siRNA knockdown efficiency rather than CAR-T cell potency, since the cells were treated with a pooled siRNA that included IFNγ-specific siRNA. No significant differences in IL-2 levels were observed between gene-specific siRNA-transfected CAR-T cells and those transfected with control siRNA (Fig. 6A), indicating preserved CAR-T cell activation.
Fig. 6: siRNA-mediated knockdown of four soluble factors in CAR-T cells does not affect CAR-T cell potency. The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.Potency of siRNA-transfected anti-CD19 CAR-T cells was assessed by cytokine secretion and cytotoxicity. A Following co-culture of siRNA-transfected anti-CD19 CAR-T cells with CD19⁺ Daudi cells at effector-to-target (E: T) ratios of 1:1 and 5:1 for 24 h, IL-2 secretion by CAR-T cells was quantified by ELISA. B Cytotoxicity of siRNA-transfected anti-CD19 CAR-T cells was assessed by measuring lysis of CD19⁺ Nalm6 target cells after 24 h of co-culture at low (1:5), medium (1:1) and high (5:1) E: T ratios. ns = not significant. Each dot represents an individual donor.
To further assess cytotoxic function, we evaluated the ability of siRNA-transfected CAR-T cells to lyse CD19⁺ Nalm6 target cells at low (1:5), medium (1:1) and high (5:1) E:T ratios. Including a low E:T ratio provides a more stringent assessment of CAR-T cytotoxic function because limited effector numbers can reveal subtle impairments that may be masked at higher ratios. No significant differences in cytotoxic activity were observed between gene-silenced and control CAR-T cells across multiple donors and various E:T ratios (Fig. 6B). Notably, we observed comparable target-cell killing at an E:T ratio of 1:5 following combined downregulation of IFN-γ, GM-CSF, GPIbα, and TNF-α, supporting the conclusion that suppressing these inflammatory mediators does not measurably compromise short-term CAR-T cytotoxic potency under in vitro conditions.
To assess whether siRNA delivery and cytokine gene silencing affect CAR-T cell fitness beyond short-term cytotoxicity, we evaluated CAR-T viability and proliferative capacity following antigen engagement. siRNA-transfected CAR-T cells were stimulated with CD19⁺ target cells and monitored over multiple time points for viability and expansion, in comparison with control siRNA-treated CAR-T cells. Under our experimental conditions, siRNA delivery and cytokine gene silencing did not significantly alter CAR-T viability, proliferation, expansion kinetics, or overall cell yield (Fig. S4).
Together, these findings demonstrate that simultaneous siRNA-mediated knockdown of key inflammatory mediators, IFN-γ, GM-CSF, TNFα, and GPIbα does not impair CAR-T cell potency in an in vitro short-term co-culture assay. These results support the feasibility of using gene silencing strategies to mitigate inflammatory toxicities without compromising the therapeutic efficacy of CAR-T cells.
Comments (0)