Sample collection
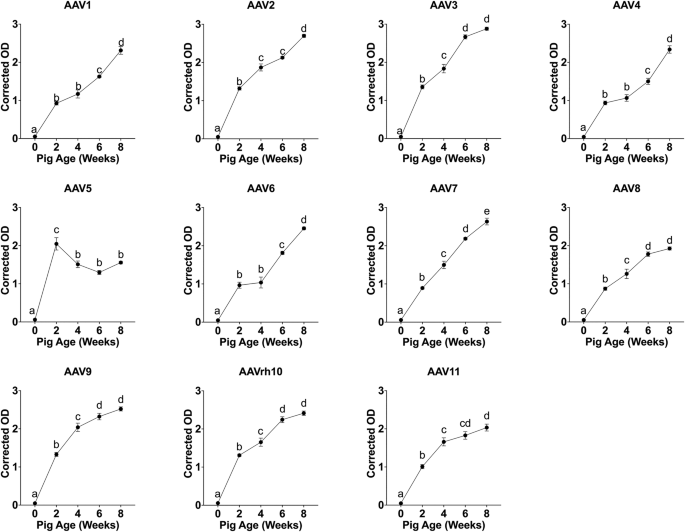
Whole blood samples were collected from independent Landrace White pigs across five developmental timepoints, 0, 2, 4, 6, and 8 weeks of age (n = 6 per age group), housed under standard facility conditions. Sample sizes were determined based on cohort availability and feasibility within the institutional swine facility. No prior power calculation was performed. Following data collection, post hoc power analysis was conducted to assess whether group sizes were sufficient to detect observed differences in antibody titers between experimental conditions.
To evaluate the influence of management practices on anti-AAV immunity, additional blood samples were obtained from 6-week-old Landrace White pigs (n = 8 per group) raised in either a standard pig housing facility or a high biosecurity facility. Animals in the high-biosecurity system were maintained under a strict “shower-in/shower-out” protocol, with personnel required to shower upon entry, and with no external clothing, footwear, or personal items permitted inside the facility. All incoming equipment and husbandry materials were cleaned extensively with disinfectants, and feed was sterilized by irradiation prior to entry. These measures were designed to minimize environmental and personnel-mediated microbial exposure, thereby providing a controlled setting to assess how reduced antigenic exposure influences the development of anti-AAV antibody responses.
Blood was collected via jugular venipuncture into serum tubes and centrifuged at 2000 rpm for 10 min at room temperature to isolate serum. Serum samples were aliquoted into labeled cryovials and stored at –20 °C until further analysis.
Enzyme-linked immunosorbent assay (ELISA)
An indirect ELISA was performed to quantify serum IgG antibodies specific to AAV capsids. High-binding polystyrene 96-well plates (Corning, Corning, NY, USA) were coated overnight at 4 °C with recombinant AAV capsids (vendors, serotypes and catalog numbers are listed in supplementary Table S1) at 1 × 10⁹ viral particles per well (i.e., 100 μL of 1 × 10¹⁰ particles/mL in PBS, pH 7.4). Plates were washed three times using a 1X wash buffer composed of 154 mM NaCl, 24.7 μM thimerosal, and 0.05% (v/v) Tween-20, delivered via a BioTek 50 | TS microplate washer (Agilent Technologies Inc., Santa Clara, CA, USA).
Wells were blocked with 5% bovine serum albumin (BSA) in PBS containing 0.05% Tween-20 (PBST) and incubated for 60 min at 25 °C with gentle rocking, consistent with standard antibody binding assays. Serum samples were diluted in blocking buffer and added to the wells, followed by a 90-min incubation under the same conditions. Because anti-AAV IgG levels differed between cohorts, higher dilutions (1:5000) were required for samples with stronger binding signals, such as those from animals housed under standard conditions. In contrast, lower dilutions (1:500) were used for cohorts with weaker signals (animals from high-biosecurity housing). The dilution factors used for the evaluations were established empirically to ensure that sample reactivities were within the linear range of the assay.
Two rabbit polyclonal antibodies served as positive controls: a broadly reactive anti-AAV VP1/VP2/VP3 antibody (Progen, Heidelberg, Germany, Cat. #61084), which recognizes multiple AAV serotypes, and an anti-AAV5 VP3 (Invitrogen, Carlsbad, CA, USA. Cat. #PA5-22817), which is predominantly reactive to AAV5 and shows weak cross-reactivity with other serotypes. Both antibodies were diluted 1:500. The anti-VP1/VP2/VP3 antibody was used to confirm capsid coating and assay functionality across all serotypes tested, whereas the anti-AAV5 VP3 antibody was included to assess serotype-restricted binding specificity. Fetal porcine serum and 5% BSA were included as negative controls.
After washing, wells were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 60 min. Goat anti-pig IgG H&L (HRP) (Abcam, Waltham, MA, USA. Cat. #ab6915) was used at 1:5000 to detect porcine IgG, and Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) (Jackson ImmunoResearch, West Grove, Pennsylvania, USA. Cat. #111-035-144) was used at the same dilution for rabbit control antibodies.
Colorimetric detection was performed using TMB substrate (Surmodics, Eden Prairie, MN, USA, Lot #TMBW240013), and reactions were stopped after 5 min using stop solution (Invitrogen, Lot #312960-000). Absorbance was measured at 450 nm using a BioTek Epoch 2 microplate reader. Raw optical density (OD) values were corrected by subtracting background absorbance from negative control wells.
Validation and dilution optimization
To ensure assay accuracy and reproducibility, we validated the ELISA platform using a monoclonal antibody standard, specificity testing, and serial dilution analysis. A standard curve generated with a purified monoclonal antibody against AAV5 VP3 showed robust linearity (R² = 0.9996), confirming the assay’s quantitative capability. Specificity testing across 11 serotypes demonstrated strong reactivity only to AAV5 with minimal cross-reactivity.
For serum dilution optimization, equal volumes of serum from eight 6-week-old pigs per facility (standard and high biosecurity) were pooled and titrated (1:10–1:50000) against AAV2 capsids. Pooling was performed solely for assay calibration to identify dilution ranges that maintained signal within the linear dynamic range of the ELISA, after which all experimental analyses were conducted on individual serum samples. Titrations were fit with a four-parameter log-logistic (4PL) model to estimate EC50. The standard-facility pool had an EC50 of 1:4 620 (95% CI in Supplementary Fig. S1), and subsequent assays were performed at 1:5000 (1.08× EC50). The high-biosecurity pool had an EC50 of 1:273 (95% CI in Supplementary Fig. S2), and assays were performed at 1:500 (1.83× EC50). These dilutions maintained OD values within the linear range and avoided saturation.
IgG depletion and validation
To determine the contribution of IgG to AAV neutralization, porcine IgG was selectively removed using Protein A/G magnetic beads (Thermo Fisher Scientific, Waltham, MA, USA. Cat. #88803). Serum from 6-week-old pigs with high anti-AAV2 reactivity was incubated with pre-washed beads at a 1:1 (v/v) ratio for 1 h at room temperature with gentle rotation. Beads were separated magnetically, and the IgG-depleted supernatant was collected.
IgG depletion was confirmed by ELISA, using plates coated with AAV2 capsids (1 × 10⁹ vg/well). Pre- and post-depletion serum samples (1:500 dilution) were probed with HRP-conjugated goat anti-pig IgG H&L antibodies (Abcam, Cat. #ab6915). A marked reduction in OD confirmed successful depletion.
For functional validation, NIH/3T3 cells were transduced with rAAV2 pre-incubated with IgG-depleted serum, untreated serum, or fetal porcine serum.
Cell culture
NIH/3T3 cells (ATCC, Manassas, VA, USA. CRL-1658) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 0.5% GlutaMAX (Gibco). Cells were maintained at 37 °C in a humidified atmosphere with 5% CO₂ and passaged at 70–80% confluency using Accutase (Innovative Cell Technologies, San Diego, CA, USA).
AAV neutralization assay
To evaluate the functional impact of pre-existing antibodies on AAV transduction, a cell-based neutralization assay was performed using NIH/3T3 cells and recombinant AAV vectors encoding green fluorescent protein (GFP). Cells were seeded in 24-well plates at a density of 0.05 × 10⁶ cells/well and cultured overnight in DMEM supplemented with 10% FBS and 0.5% GlutaMAX.
Recombinant AAV2, AAV6, AAV8, AAV9, and AAV11 vectors (Table S1) were applied at a fixed multiplicity of infection (MOI) of 1 × 10³ vector genomes (vg) per cell. This constant MOI was used to standardize viral genome input across serotypes. Viral titers were confirmed by genome copy quantification prior to use. No adjustments were made to equalize transduction output (e.g., GFP expression) between serotypes, as differences in baseline transduction efficiency reflect intrinsic capsid properties and were accounted for through normalization during the assessment of antibody-mediated inhibition.
AAV5 was excluded from the neutralization analyses because robust GFP expression could not be achieved under these standardized conditions. As normalization of transduction required a measurable baseline signal, the absence of reliable GFP expression precluded accurate quantification of serum-mediated inhibition for this serotype.
Porcine serum samples were diluted in FBS at ratios of 1:0 (porcine serum only), 1:9, 1:4, 2:3, and 0:1 (FBS only), and incubated with each tested rAAV for 1 h at 37 °C to allow antibody binding under physiologically relevant conditions. Virus–serum mixtures were then added to cells in triplicate wells.
After 48 h of incubation at 37 °C and 5% CO₂, cells were harvested using Accutase and analyzed for GFP expression by flow cytometry (Invitrogen Bigfoot Cell Sorter, Thermo Fisher Scientific). Transduction efficiency was quantified as the percentage of GFP-positive cells relative to total events.
For each serotype, a no-serum control condition (0:1) was used to establish baseline transduction. Transduction in the presence of serum was normalized to this control, which was set to 100%, and calculated as:
Relative Transduction (%) = (GFP-positive cells in serum-treated condition ÷ GFP-positive cells in no-serum control) × 100
This normalization strategy enabled comparison of the relative inhibitory effects of serum antibodies across serotypes independent of intrinsic differences in capsid-mediated transduction efficiency. Because porcine serum contains polyclonal antibodies with variable affinity and avidity for distinct capsid epitopes, normalization to a matched no-serum control ensured that observed reductions in transduction reflected antibody-mediated inhibition rather than differences in viral input or baseline infectivity.
Flow cytometry acquisition and analysis
At the specified time post-transduction, cells were washed with PBS, dissociated to a single-cell suspension using Accutase, and resuspended in flow buffer (PBS supplemented with 1% FBS). Samples were acquired on an Invitrogen Bigfoot Cell Sorter (Thermo Fisher Scientific) using a 488-nm laser, and GFP fluorescence was collected on the FITC channel. Non-transduced cells were used to define background autofluorescence and establish the GFP-positive threshold; single-stained controls were used for compensation when applicable. At least 10000 events were collected per sample.
Flow cytometry data were analyzed using the instrument’s analysis software. Gating was performed sequentially to exclude debris based on forward and side scatter, followed by visualization of single-cell events using FSC height versus FSC area. GFP-positive and GFP-negative populations were defined based on fluorescence intensity using SSC versus GFP dot plots and confirmed by one-dimensional GFP histograms. Because the analysis was focused on quantifying transduction efficiency rather than cell sorting, singlet discrimination was not strictly enforced to maximize the number of events analyzed.
Sequence retrieval and multiple sequence alignment
AAV capsid protein sequences for serotypes AAV1 through AAV9 and AAVrh10 were retrieved from the UniProt Knowledgebase (UniProtKB) using the following accession numbers: AAV1 (Q9WBP8), AAV2 (Q87087), AAV3 (QDH44565.1), AAV4 (Q9J4G2), AAV5 (Q9JJZ2), AAV6 (Q9J4G0), AAV7 (Q8BHD3), AAV8 (Q8BHD2), AAV9 (Q8BHD1), and AAVrh10 (G3UUT3). Full-length amino acid sequences were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/), with manual segmentation into the VP1 unique region (VP1u), VP1/VP2 common region, and VP3 region. AAV11 was excluded from the sequence identity analysis due to the lack of a reliable, full-length capsid sequence suitable for comparative alignment.
Pairwise sequence identity analysis and visualization
Alignments were processed in R (version 4.3.1) using the Biostrings package to compute pairwise sequence identity matrices. Heatmaps were generated using the pheatmap package and hierarchically clustered to assess relationships among AAV serotypes. Sequence identity was evaluated for full-length capsids and subdomains to assess correlation with serological cross-reactivity.
In parallel, ELISA antibody reactivity profiles were compiled for each serotype across all serum samples, and pairwise Pearson correlation coefficients between serotypes were calculated based on these reactivity profiles. Pairwise sequence identity matrices were then compared with the corresponding serological correlation matrices to assess the relationship between capsid similarity and antibody cross-reactivity.
Statistical analysis
All statistical analyses were conducted in R (version 4.3.1). Linear mixed-effects models (LMMs) were used to evaluate the effects of Age, Serotype, and Facility on ELISA OD values and neutralization outcomes using the lme4 package. Random effects included Pig_ID (individual variation within and across litters) and/or Sow_ID (maternal influence), depending on design.
To evaluate differences in antibody levels across developmental stages and serotypes, LMMs were fitted with age and serotype as fixed effects and pig identity nested within sow identity as random effects. Because the nested sow-level effect was estimated near zero and produced singular fits, the final model used a pig-level random intercept. The model was specified as: Corrected_ODijkl = µ + αi + βj + (αβ)ij + µk + ϵijkl, where µ is the overall mean, αi is the fixed effect of age, βj is the fixed effect of serotype, (αβ)ij is their interaction, µk ~ N(0,σ²) is the pig-level random intercept, and ϵijkl is the residual error. Post hoc pairwise comparisons were performed using estimated marginal means with Tukey adjustment for multiple comparisons.
Model assumptions were evaluated through residual diagnostics. Homogeneity of variance was assessed by visual inspection of residual-versus-fitted plots and by Levene’s tests across serotype groups, which did not detect statistically significant variance differences. Simulation-based diagnostics using the DHARMa package further supported appropriate dispersion. Sensitivity analyses using log-transformed ELISA values yielded consistent results, indicating that conclusions were robust to potential mild heteroscedasticity.
Variance component analysis was done using restricted maximum likelihood (REML). Simulation-based power analyses were conducted using the simr package (nsim = 100) for two scenarios: (1) detecting Age × Serotype interactions using longitudinal ELISA data (n = 30 pigs across five ages); and (2) detecting inter-facility differences in serotype reactivity at 6 weeks of age (n = 16).
Pearson correlation coefficients (r) were used to assess the relationship between ELISA OD values and AAV transduction efficiency. Correlation matrices and visualizations were generated using ggplot2 and pheatmap.
Unless otherwise noted, results are presented as mean ± standard error of the mean (SEM), with α = 0.05 considered statistically significant.

Comments (0)