
Remember me




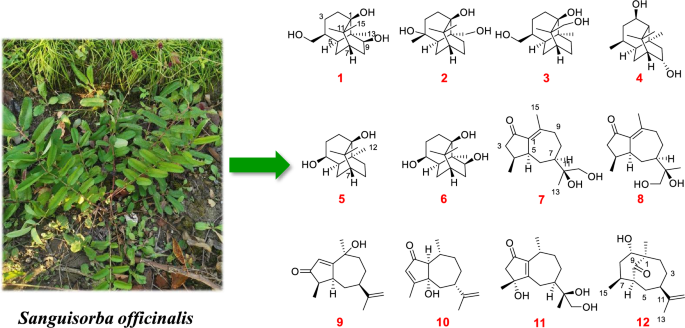
Compound 1 was isolated as colorless needle crystals (MeOH), assigned the molecular formula C15H26O3 according to the HRESIMS ion peak at m/z 277.1775 [M + Na]+ (calcd for C15H26O3Na, 277.1774), requiring an index of hydrogen deficiency (IHD) of three. The 1H NMR data showed an O-bearing methine at δH 3.56 (1H, brd, J = 8.9 Hz, H-9), a pair of oxygenated methylene at 3.38 (1H, dd, J = 10.7, 8.0 Hz, H-12a) and 3.35 (1H, dd, J = 10.7, 6.5 Hz, H-12b), three methyl singlets at δH 1.11 (3H, s, Me-13), 1.16 (3H, s, Me-14) and 1.17 (3H, s, Me-15), and multiple methylene and methine signals in high fields δH 1.22–2.13 (Table 1). In its 13C and DEPT-135 NMR spectra (Table 2), fifteen carbons were observed including three quaternary carbons containing an oxygenated carbon at δC 78.4 (C-1), four methine carbons with an O-bearing at δC 76.7 (C-9), five methylene carbons including an oxygenated one at δC 65.4 (C-12), as well as three methyl carbons at δC 18.2 (C-13), 25.1 (C-14), 27.5 (C-15). The above 13C NMR profile showed close agreement with that of the known patchoulol-type sesquiterpenoid, (9R)-9-hydroxypatchoulol [21]. Notably, compound 1 featured a hydroxymethyl group at C-4 [δC 65.4 (C-12), δH 3.38 (1H, dd, J = 10.7, 8.0 Hz, H-12a), 3.35 (1H, dd, J = 10.7, 6.5 Hz, H-12b)], whereas the known analogue (9R)-9-hydroxypatchoulol is substituted with a methyl group at C-12. The proposed structure of 1 was consistent with its molecular formula. Key HMBC correlations from H2-12 to C-3, C-4, and C-5 positioned the hydroxyl group at C-12 (Fig. 2). Furthermore, the contiguous proton spin system of H2-2/H2-3/H-4/H-5(H2-12)/H2-6/H-7/H2-8/H-9 observed in the 1H–1H COSY spectrum (Fig. 2), unequivocally confirmed the planar framework of 1. The relative configuration of 1 was assigned based on NOESY correlations in conjunction with biosynthetic analogy. Within patchoulol-type sesquiterpenoids isolated from Pogostemon cablin, Me-13 commonly adopts α-oriented [21, 22]. The NOESY spectra (Fig. 3) of Me-13/H-4/H-5/H-9 supported the assignment H-4, H-5, and H-9 as α-oriented. Therefore, both of 9-OH and 12-CH2OH adopt a β-orientation. Finally, the absolute configuration of 1 was unambiguously defined as (1R,4S,5S,7S,9S,10R) [Cu-Kα radiation, Flack parameter = 0.05(11), CCDC 2,333,284, Fig. 4] by the X-ray crystallographic analysis. Thus, compound 1 was identified as (1R,4S,5S,7S,9S,10R)-9,12-dihydroxypatchoulol, named sanguisorbaol A.
Table 1 1H NMR data of compounds 1–12 in CD3OD (δ in ppm, J in Hz, measured at 600 MHz)Table 2 13C NMR data of compounds 1–12 in CD3OD (δ in ppm, measured at 150 MHz)Fig. 2
Key 1H–1H COSY and HMBC correlations of compounds 1–12
Fig. 3
Key NOESY correlations of compounds 1–12
Fig. 4
X‑ray crystal structures of compounds 1, 3, 5 and 12
Obtained as a colorless oil, compound 2 exhibited the same molecular formula as 1, as evidenced by its HRESIMS peak at m/z 277.1775 [M + Na]+ (calcd for C15H26O3Na, 277.1774). This formula corresponded to three IHD. The 1H NMR data showed a pair of oxidized methylene protons at δH 4.00 (1H, d, J = 11.4 Hz, H-13a) and 3.50 (1H, d, J = 11.4 Hz, H-13b), three methyls at δH 1.17 (3H, s, Me-12), 1.07 (3H, s, Me-14) and 1.09 (3H, s, Me-15), as well as multiple methylene and methine signals in high fields δH 1.19–2.01 (Table 1). The 15 carbons signals in the 13C NMR spectrum and HSQC of 2 were classified as four fully substituted carbons atoms [including two oxygenated at δC 77.3 (C-1) and 74.2 (C-4)], two methine, six methylene [including one oxygenated at δC 68.8 (C-13)] and three methyls [δC 28.9 (C-12), 27.5 (C-14), 24.5 (C-15)] (Table 2). Comparative 1D NMR spectra revealed that 2 was structurally analogous to (4R*)-4 hydroxypatchoulol [22]. The principal distinction between 2 and the known analogue lies in the substitution of a methyl group by an oxygenated methylene at δC 68.8. The assignment of the hydroxymethyl at C-10 was corroborated by HMBC analysis of δH 4.00 (1H, d, J = 11.4 Hz, H-13a), 3.50 (1H, d, J = 11.4 Hz, H-13b) to δC 77.3 (C-1), 44.8 (C-5), 25.6 (C-9) and 42.3 (C-10) (Fig. 2). The relative configuration of 2 was established by virtue of NOESY correlation (Fig. 3). The observed correlation between H-5 and H2-13 indicated that both are α-oriented, whereas no correlative signal was detected between H2-13 and Me-12 suggested a β-orientation for Me-12. According to the calculated ECD spectral data (Fig. 5) together with the consistent biosynthetic pathway (Fig. 6), the absolute configuration of 2 was determined to be 1R,4R,5R,7R,10S. Consequently, the structure of 2 was established and designated sanguisorbaol B.
Fig. 5
The experimental and calculated ECD spectra of compounds 1–12
Fig. 6
Two hypothetical biosynthetic pathways of patchoulol- and guaiane-type sesquiterpenoids in S. officinalis (radical mechanisms on the left and ionic mechanisms on the right)
Colorless needle crystals of compound 3 were isolated from MeOH, and its molecular formula C15H26O3 was corroborated by the quasi-molecular ion peak at m/z 277.1773 [M + Na]+ (calcd for C15H26O3Na, 277.1774) in HREIMS. The 1H NMR data (Table 1) displayed two pairs of O-bearing methylene protons at δH 3.38 (1H, dd, J = 10.7, 8.0 Hz, H-12a) and 3.34 (1H, dd, J = 10.7, 6.7 Hz, H-12b), 3.95 (1H, d, J = 10.7 Hz, H-15a) and 3.36 (1H, d, J = 10.7 Hz, H-15b), two methyl singlets at δH 0.87 (3H, s, Me-13) and 1.18 (3H, s, Me-14), and multiple methylene and methine signals in high fields δH 1.09–2.03. Its 13C NMR spectrum displayed 15 carbons combined with DEPT-135 corresponding to three quaternary carbon atoms, three methine carbons, seven methylene, and two methyls. Among them, δC 78.1 (C-1), 65.3 (C-12), 70.8 (C-15) were three oxygenated carbons. The 1H and 13C NMR spectra of compound 3 closely resembled those of the known compound 19 [23], with the exception of the characteristic substitution of a singlet methyl (C-12) by a hydroxymethyl group in 3 (Tables 1 and 2). Key HMBC corrections from H2-12 (δH 3.38 and 3.35) to C-3 (δC 24.4), C-4 (δC 37.5) and C-5 (δC 39.6), and H2-15 (δH 3.95 and 3.36) to C-1 (δC 78.1), C-7 (δC 36.5), C-11 (δC 44.9) and C-14 (δC 18.9), could confirm that two hydroxyl groups were connected at C-12 and C-15, respectively (Fig. 2). NOESY correlations (Fig. 3) of H-7/Me-14/H2-15 and Me-13/H-4/H-5 revealed H-4, H-5 and Me-13 were α-oriented, while H-7 and H2-12 were β-oriented, consistent with the commonly observed in patchoulol-type sesquiterpenoids. In addition, NOESY correlations of Me-14 (δH 1.17) and H-6β (δH 1.36), H2-15 (δH 3.95 and 3.36) and H-9β (δH 2.03) demonstrated that Me-14 and C-6 were situated on the same face, while CH2-15 and C-9 were located on the opposite face. Finally, direct verification via X-ray crystallography established the absolute configuration of compound 3 as (1S,4S,5S,7R,10S,11S) [Cu Kα radiation, Flack parameter = 0.10(11), CCDC 2,333,293, Fig. 4]. Hence, the structure of 3 was finally determined and named as sanguisorbaol C.
Compound 4 was isolated as a colorless oil, possessing the molecular formula C15H26O3 relying on the peak signal at m/z 237.1833 [M − H2O + H]+ (calcd for C15H25O2, 237.1849) from HRESIMS, with an IHD of three. By meticulously comparing the 1D NMR spectroscopic data of 4 with those of 3α,8α-dihydroxypathoulol [21], the two compounds were found to be structurally analogous (Tables 1 and 2). The major differences between them lie in the down-field shift of C-2 (δC 72.2 vs. 42.7) and the up-field shifts of C-3 (δC 36.8 vs. 72.9) and C-4 (δC 25.7 vs. 37.7), suggesting the transfer of the hydroxyl group from C-3 to C-2 in 4, as further verified via the HMBC corrections from H-2 (δH 3.81) to C-1 (δC 75.8), C-3 (δC 36.8), C-4 (δC 25.7), C-10 (δC 39.6) and C-11 (δC 40.9) (Fig. 2). In addation, the correlations (Fig. 2) of H-2/H2-3/H-4/H-5(Me-12)/H2-6/H-7/H-8/H2-9 in the 1H–1H COSY spectrum unambiguously confirmed the planar framework of 4. Correlations of H-5/H-4/Me-13 in the NOESY spectrum (Fig. 3) suggested these protons were cofacial, and were assigned an α-orientation. In contrast, NOESY interactions among H-7/H-8/Me-14/Me-15 and H-2/Me-14 suggested that these protons were β-orientation, and the 2-OH and 8-OH in 4 were α-orientation. Finally, the absolute configuration of 4 was established as 1R,2S,4S,5S,7R,8S,10S, based on the shared biosynthetic pathway (Fig. 6) and supporting evidence from ECD calculations (Fig. 5). Thus, the compound 4 was determined to be (1R,2S,4S,5S,7R,8S,10S)-2,8-dihydroxypatchoulol and named trivially as sanguisorbaol D.
Colorless needle-like crystals of compound 5 were purified from EtOH, possessing the molecular formula C14H24O2 according to the HRESIMS ion peak at m/z 207.1742 [M − H2O + H]+ (calcd for C14H23O, 207.1743), and corresponding to an IHD of three. Analysis of its 1H NMR spectrum (Table 1) exhibited an oxygenated methine signal at δH 4.08 (1H, ddd, J = 10.3, 6.2, 3.7 Hz, H-4), and three methyls at δH 0.88 (3H, s, C-12), 1.14 (3H, s, C-13) and 1.08 (3H, s, C-14). In combination with the DEPT spectrum, the 13C NMR data (Table 2) revealed 14 carbon resonances, which included three quaternary carbons [one oxygenated at δC 76.3 (C-1)], three methine carbons [one oxygenated at δC 68.4 (C-4)], five methylenes and three methyls [δC 21.3 (C-12), 24.7 (C-13), 27.7 (C-14)]. The above 1D NMR data showed some features analogous to those of 24 [24], apart from the substitution of the α,β-unsaturated ketone moiety by two methylenes and one oxygenated methine group in 5. The above analyses suggested that compound 5 belongs to the nor-patchoulol-type sesquiterpenoid skeleton. The key HMBC correlations from H-4 to C-2, C-3, C-5, C-6 and C-10 additionally confirmed the absence of -CH3 and the attachment of a hydroxyl group at C-4 (Fig. 2), with the assistance of 1H–1H COSY correlations of H2-2/H2-3/H-4/H-5/H2-6/H-7/H2-8/H2-9. The NOESY spectrum of Me-12/H-4/H-5 (Fig. 3) implied that H-4, H-5 along with Me-12 were α-orientations, and 4-OH was β-orientation. In the X-ray diffraction experiment [Cu-Kα radiation, Flack parameter = 0.00(8), CCDC 2,333,300], the (1R,4S,5R,7R,10S) absolute configuration was established as 5 (Fig. 4). Thus, the structure of 5 was determined as (1R,4S,5R,7R,10S)-4-hydroxy-nor-patchoulol and designated as sanguisorbaol E.
Compound 6 featured the molecular formula of C14H24O3 (IHD of three) determined by HRESIMS at m/z 263.1617 [M + Na]+ (calcd for C14H24O3Na, 263.1618). Thorough investigation of the 1H and 13C NMR of 6 indicated that it had a structural similarity to 5, apart from an extra hydroxyl group in 6 (Tables 1 and 2), which was in line with the increase in molecular weight of 6 by 16-units. The key HMBC cross-peaks of H-9 with C-1, C-5, C-7, C-8, C-10 and C-12, as well as the 1H–1H COSY correlations of H2-2/H2-3/H-4/H-5/H2-6/H-7/H2-8/H-9, located the other hydroxyl group at C-9 (Fig. 2). In the NOESY spectrum (Fig. 3), the correlation from H-9 to H-5 and Me-12 suggested the 9-OH was β-orientation. Guided by the shared biosynthetic pathway (Fig. 6) and supported by ECD calculations (Fig. 5), the absolute configuration of 6 was elucidated as 1R,4S,5R,7S,9S,10R. In consequence, the structure was characterized as (1R,4S,5R,7S,9S,10R)-4,9-dihydroxy-nor-patchoulol and designated as sanguisorbaol F.
Compounds 7 and 8 were both colorless oils. Their molecular formulas were determined using HRESIMS to be C15H24O3 (IHD of four) at m/z 253.1798, and 253.1800 ([M + H]+ calcd for C15H25O3, 253.1798), respectively. The 1H NMR signals (Table 1) of 7 displayed a pair of oxygenated methylene signals at δH 3.50 (1H, d, J = 11.2 Hz, H-12a) and 3.44 (1H, d, J = 11.2 Hz, H-12b), three methyl signals at δH 1.06 (3H, s, H-13), 0.98 (3H, d, J = 6.9 Hz, H-14) and 2.23 (3H, d, J = 2.1 Hz, H-15). Its 13C NMR (Table 2) together with DEPT data showed 15 carbon resonances attributable to a carbonyl quaternary carbon at δC 209.9 (C-2), two olefinic carbons at δC 138.2 (C-1) and 157.4 (C-10), an oxygenated quaternary carbon at δC 76.1 (C-11), three methines, five methylenes containing an O-bearing one at δC 68.9 (C-12), and three methyls at δC 20.9 (C-13), 16.3 (C-14) and 21.7 (C-15). Detailed examination of 1D NMR data showed that the framework of 7 was close to the known guaiane-type sesquiterpenoid xylaguaianol D [25], only differing by incorporation of a carbonyl quaternary carbon (δC 209.9) in 7 instead of one methylene carbon in xylaguaianol D. The placement of the carbonyl at C-2 was corroborated by the HMBC correlations (Fig. 2) of H2-3 with C-2, C-4, C-5 and C-14, of Me-15 with C-1, C-2, C-9 and C-10. Besides, key HMBC correlations of Me-13 with C-7, C-11 and C-12 and 1H–1H COSY correlations of H2-3/H-4/H-5(H3-14)/H2-6/H-7/H2-8/H2-9 (Fig. 2) unequivocally confirmed the planar structure of 7. Correlations of H-4/H-5/H-6α (δH 2.09)/H-7 and Me-14/H-6β (δH 1.09)/Me-13 in the NOESY spectrum (Fig. 3) revealed the coplanarity of H-4, H-5 and H-7, which were arbitrarily designated as having an α-orientation, while Me-15 exhibited a β-orientation.
The planar structure of 8 was found to closely resemble that of 7 through comparing the 1H and 13C NMR spectra. By careful comparison of the 13C NMR (Table 2) signals between 7 and 8, subtle differences were observed at C-6 (δC 29.0 for 7, δC 30.2 for 8) and C-8 (δC 27.5 for 7, δC 26.4 for 8), implying that 7 and 8 formed a pair of R/S epimers at C-11 position. Key NOESY correlations of H-12/H-8 and Me-13/H-6 in 7 implied that C-12 and C-8 were on the same side, and Me-13 and C-6 on the other side. On the contrary, the NOESY correlations of H-12/H-6 and Me-13/H-8 in 8 implied that C-12 and C-6 were on the same side, and Me-13 and C-8 on the other side (Fig. 3). This enabled us to distinguish the 11S* configuration for 7 and the 11R* for 8 by assuming 7R-configuration for both compounds. Subsequently, four molecular models (4S,5S,7R,11S)-7a, (4R,5R,7S,11R)-7b, (4S,5S,7R,11R)-8a, and (4R,5R,7S,11S)-8b were established and ECD calculations were performed. Subsequently, compared with experimental ECD spectra, it was found that compounds 7 and 8 matched well with (4S,5S,7R,11S)-7a and (4S,5S,7R,11R)-8a. Based on the excellent consistency between the calculated ECD spectrum and the experimental one (Fig. 5), the absolute configuration of compounds 7 and 8 were ascertained to be (4S,5S,7R,11S) and (4S,5S,7R,11R), respectively. Accordingly, the complete structures of compound 7 and 8 were fully identified as (4S,5S,7R,11S)-4α,5α,7α(H)-2-oxo-1(10)-guaiaene-11,12-diol and (4S,5S,7R,11R)-4α,5α,7α(H)-2-oxo-1(10)-guaiaene-11,12-diol, and named as sanguisorbaol G and sanguisorbaol H, respectively.
Compound 9 was afforded as colorless oil with the molecular formula C15H22O2 deduced from HRESIMS at m/z 235.1696 [M + H]+ (calcd for m/z C15H23O2, 235.1693), implying an IHD of five. Detailed analysis of NMR spectrum (Tables 1, 2 and Fig. 2) demonstrated the structure of 9 closely parallels that of stelleranoid L [26], differing only in the relative configuration at C-5. Key NOESY correlations between H-5 and H-4, H-7 and Me-15 revealed that H-4, H-5, H-7 and Me-15 were placed on the same side of the [5, 7] bicyclic guaiane-type sesquiterpenoid and assigned as α-orientation, while the Me-14, isopropenyl-7 and 10-OH were β-orientation in 9 (Fig. 3). Ultimately, the absolute configuration of 9 was ascertained as (4R,5S,7R,10S) via analysis of theoretical and experimental ECD curves (Fig. 5), and 9 was thus trivially named as sanguisorbaol I.
Compound 10, a colorless oil, had its molecular formula determined to be C15H22O2 supported by the ion peak at m/z 235.1692 [M + H]+ (calcd for C15H23O2, 235.1693) from HRESIMS, with an IHD of five. The NMR spectroscopic data of 10 (Fig. 2, Table 1 and 2) showed it shared the same planar structure with 2-keto-5-hydroxyguai-3,11-diene [27], along with the main variation was ascribed to the C-7 relative configuration. Key NOESY correlations (Fig. 3) of H-1/Me-15 and H-7/H-10 suggested that H-1 and Me-15 occupied cofacial and were designated as α-oriented, while H-7 was β-oriented. Additionally, key NOESY correlations of 5-OH [δH 5.13 (1H, s)]/H-1/H-3 in the DMSO-d6 determined that 5-OH was α-orientation. Ultimately, the measured ECD spectrum of 10 (Fig. 5) was consistent with the computed ECD spectrum for (1S,5S,7S,10R), thus determining the absolute configuration of 10 as delineated. Eventually, the structure of 10 was established as (1S,5S,7S,10R)-5α-hydroxy-1α,7β,10β,(H)-guaia-3(4),11(12)-dien-2-oxo and named as sanguisorbaol J.
The molecular formula of 11 (colorless oil) was deduced to be C15H24O4 from its HRESIMS ions at m/z 269.1746 [M + H]+ (calcd for C15H25O4, 269.1747) together with 13C NMR spectroscopic signals, requiring an IHD of four. Comparative analysis of the NMR spectral info (Fig. 2, Table 1 and 2) of compound 11 with (4R,7S,10S)-2-oxo-4α,11-dihydroxyguaia-1(5)-ene revealed substantial similarities [28], with the exception that one oxygenated methylene group in 11 replaced the methyl group. These observations were supported by the HMBC correlations (Fig. 2) from Me-13 to C-7, C-11 and C-12, and located the oxygenated methylene at C-12. The NOESY correlations (Fig. 3) of Me-15/H-6β (δH 2.24)/H-7 indicated that H-7 and Me-15 were β-oriented, since the NOESY cross-peaks of Me-14/H-6α (δH 3.08) confirmed these protons were α-oriented. However, it was not feasible to determine the relative configuration at C-11 based on the above data. In response to the uncertainty of C-11, four possible isomers models, namely, (4S*,7S*,10S*,11S*), (4S*,7S*,10S*,11R*), (4R*,7R*,10R*,11R*) and (4R*,7R*,10R*,11S*), which constitute a pair of epimers and their enantiomers. Two epimers (4S*,7S*,10S*,11S*)-1 and (4S*,7S*,10S*,11R*)-2 underwent GIAO based quantum chemical 1H and 13C NMR calculation at MPW1PW91/6-311G(d,p) level utilizing the solvation model based on density (SMD) in methanol. Then, the DP4 + analysis was performed and the results (Figure S5–101) showed that (4S*,7S*,10S*,11R*)-2 (99.90%) was significantly better than (4S*,7S*,10S*,11S*)-1 (0.10%), which could be determined the relative configuration of 11 was (4S*,7S*,10S*,11R*) or (4R*,7R*,10R*,11S*). Eventually, the absolute configuration of compound 11 was established as (4R,7R,10R,11S) through analysis of the theoretical and measured ECD curves (Fig. 5), and 11 was elucidated as (4R,7R,10R,11S)-7α,10β,(H)-2-oxo-1(5)-guaiaene-4α,11,12-triol and designated as sanguisorbaol K.
Compound 12 was purified as colorless needle crystals (MeOH). The molecular formula of compound 12 was assigned as C15H24O2 according to the HRESIMS at m/z 237.1847 [M + H]+ (calcd for C15H25O2, 237.1849), with four IHD. The 1H NMR data (Table 1) exhibited a pair of olefinic protons at δH 4.63 (1H, br s, H-12a) and 4.58 (1H, br s, H-12b), one O-bearing methine at δH 3.81 (1H, t, J = 2.7 Hz, H-9), and three methyls at δH 1.68 (3H, br s, H-13), 1.03 (3H, s, H-14) and 1.00 (3H, d, J = 6.7 Hz, H-15). A detailed analysis of the 13C NMR (Table 2) and DEPT spectrum exhibited 15 carbon resonances, encompassing three quaternary carbons containing a carbonyl group at δC 220.2 (C-10) and an olefinic one at δC 152.4 (C-11), four methines with an oxygenated one at δC 78.5 (C-9), five methylenes with an olefinic carbon at δC 109.0 (C-12), and three methyls. Among which, δC 152.4 (C-11), 109.0 (C-12) and 19.5 (C-13) indicated the presence of an isopropenyl unit in the structure. Taking into account that two IHDs were ascribed to the carbonyl group and the double bond, 12 should also be a bicyclic sesquiterpene. The 1H–1H COSY correlation of H2-2/H2-3/H-4/H2-5/H-6/H-7/H2-8(Me-15)/H-9, complemented by a series of HMBC correlations (Fig. 2) from Me-14 to C-1, C-2, C-9 and C-10, H-6 to C-1, C-4, C-5, C-7, C-8, C-10 and C-15, and H2-2, H2-5, H-9 to C-10, unambiguously confirmed a 6/7-fused bicyclic framework featuring C-1 and C-6 as bridgehead carbons and the carbonyl group located on the bridge. Additionally, the HMBC correlations from Me-15 to C-6, C-7, and C-8 indicated that Me-15 was attached to C-7. Simultaneously, the HMBC correlations of Me-13 with C-4, C-11, and C-12, combined with those of H2-12 with C-4, C-11, and C-13 revealed the presence of an isopropenyl at C-4. Moreover, C-9 was deduced to be functionalized with a hydroxyl group based on its oxygenated characteristic and molecular formula. The NOESY correlations (Fig. 3) of H-4 with H-6, H-9 with Me-15, and H-9 with H2-2 demonstrated that H-4, H-6, H-7, Me-14, and 9-OH adopted a uniform orientation and assigned as α, thereby disclosing that the bridged carbonyl group resided beneath the bicyclic framework. Thereafter, ECD simulations were employed to speculate the absolute stereochemistry of 12. The calculated ECD spectrum of (1R,4R,6S,7S,9S)-12a matched well with the experimental one (Fig. 5), confirmed by X-ray crystallography experiment [Cu-Kα radiation, Flack parameter = 0.01(10), CCDC 2,333,292] (Fig. 4). As a result, the structure of 12 was elucidated and given the name sanguisorbaol L. Structurally, 12 was the third natural occurring isopropenyl-branched bicyclo [4.3.1] decanone sesquiterpenoid featuring an angular methyl and a secondary methyl, following pogocablenes A and B from P. cablin [24]. The absolute configuration was first definitively identified using single-crystal X-ray diffraction technology.
In addition, twenty-five known compounds (13–37) were isolated from S. officinalis. Their structures were determined to be (3R*)-3 hydroxypatchoulol [22] (13), (5R)-5-hydroxypatchoulol [29] (14), 6β-hydroxypatchoulol [21] (15), (8S*)-8 hydroxypatchoulol [22] (16), (9R*)-9 hydroxypatchoulol [22] (17), 1,3,4,4α,5,6,7,8α-octahydro-1-hydroxy-4,8α,9,9-tertramethyl-1,6-methanonaphthalen-8-(2H)one [30] (18), ( ±)-hydroxy patchouli alcohol [23] (19), 3α,9β-dihydroxypathoulol [23] (20), (5R,8S)-5,8 dihydroxypatchoulol [22] (21), (6S*,9S*)-6,9 dihydroxypatchoulol [22] (22), 8α,9α-dihydroxypatchoulol [21] (23), pogocablene M [24] (24), 2-keto-1(5)-β-patchoulene-4β-ol [31] (25), 2-keto-1(5)-β-patchoulene-4α-ol [31] (26), (3R,5R,8S)-3-hydroxy-3,8-dimethyl-5-(prop-1-en-2-yl)-3,4,5,6,7,8-hexahydroazulen-1(2H)-one [32] (27), (3S,5R,8S)-3-hydroxy-3,8-dimethyl-5-(prop-1-en-2-yl)-3,4,5,6,7,8-hexahydroazulen-1(2H)-one [32] (28), pogocablene J [24] (29), (1R,4S,5S,7R,10R,11R)-guaiane-10,11,12-triol [33] (30), xylaguaianol C [25] (31), pogocablene O [24] (32), clovane-2β,9α-diol [34, 35] (33), senecrassidiol [36] (34), caryolane-1,9β-diol [34] (35), linariophyllene A [
Comments (0)