3.1 General experimental protocols
All reactions conducted at temperatures higher than the ambient laboratory temperature were carried out in a silicone oil bath, which was preheated to the target temperature before the reaction vessels were immersed in it. Anhydrous solvents were purified and dried according to standard procedures, while commercially available reagents were used without further purification. Column chromatography was typically performed using silica gel (200–300 mesh, Qingdao Marine Chemical Co. Ltd., Qingdao, People’s Republic of China) as the stationary phase. The progress of the reactions was monitored in real time by thin-layer chromatography (TLC), with the reaction progress observed under ultraviolet light and stained with phosphomolybdic acid in ethanol solution and heat. Petroleum ether: ethyl acetate was used as the eluent. IKA ElectraSyn 2.0 electrolysis devise was used as electrolytic instrument, which was equiped graphite plates electrode (5.2 cm × 0.8 cm × 0.2 cm) as anode and cathode.
Optical rotations were measured using a Jasco P-1020 polarimeter equipped with a 1 dm pathlength cell.1H and 13C NMR spectra were recorded with tetramethylsilane as an internal standard at ambient temperature with Zhongke 400 MHz or Bruker 600 MHz NMR spectrometer in CDCl3 or CD3OD unless specified. Splitting patterns are designated as singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), quintet (quin), doublet of doublets (dd), and triplet of doublets (td). Splitting patterns that cannot be interpreted or easily visualized are designated multiplet (m). HRMS data were recorded via positive ion electrospray or electron impact mass spectrometry using a time of flight analyzer. Experimental ECD spectra were measured on a Chirascan instrument. Chiral semi-preparative HPLC was performed on an Agilent 1100 HPLC with the CHIRALPAK® AD-H (4.6 mm × 250 mm) column.
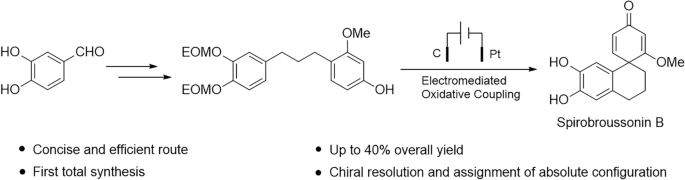
3.1.1 Synthesis of 3,4-bis(ethoxymethoxy)benzaldehyde, 2
To a dried reaction flask charged with 3,4-dihydroxybenzaldehyde (1.40 g, 10.0 mmol) in CH2Cl2 (50 mL) were sequentially added 4-dimethylaminopyridine (122 mg, 1.00 mmol), N, N-diisopropylethylamine (5.20 g, 40.0 mmol), and chloromethyl ethyl ether (3.80 g, 40.0 mmol). The flask was fitted with a drying tube, and the reaction mixture was stirred at room temperature for 7 h. Upon completion of the reaction as monitored by TLC (disappearance of the starting material), the mixture was quenched with water and extracted with CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification by silica gel column chromatography (petroleum ether/ethyl acetate, 15:1, v/v) afforded compound 2 as a yellow–brown oil (2.35 g, 92.5% yield). 1H NMR (600 MHz, CDCl3) δ: 9.86 (s, 1H), 7.68 (d, J = 2.0 Hz, 1H), 7.50 (dd, J = 8.3, 2.0 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 5.36 (s, 2H), 5.32 (s, 2H), 3.80–3.74 (m, 5H), 1.22 (td, J = 7.1, 3.8 Hz, 7H). 13C NMR (150 MHz, CDCl3) δ: 191.0, 152.9, 147.6, 131.1, 126.3, 116.0, 115.4, 94.1, 93.7, 64.9, 64.8, 15.2, 15.1. HRMS (ESI) m/z: [M + H]+ calcd. for [C13H18O5 + H]+ 255.2901, found: 255.1235.
3.1.2 Synthesis of (E)-3-[3,4-bis(ethoxymethoxy)phenyl]-1-(4-hydroxy-2-methoxyphenyl)prop-2-en-1-one, 4
Compound 2 (1.27 g, 5.0 mmol) was dissolved in EtOH (40 mL) in a dried reaction flask, followed by the addition of 1-(4-hydroxy-2-methoxyphenyl)ethan-1-one (830 mg, 5.0 mmol) and potassium hydroxide (2.8 g, 50 mmol). The reaction mixture was stirred at room temperature for 1.5 h, at which point TLC analysis indicated the almost complete consumption of the starting material. The reaction was then quenched by adjusting the pH to neutrality and adding water. The resulting mixture was concentrated under reduced pressure to remove EtOH, followed by extraction with CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford the crude product. Purification by silica gel column chromatography (petroleum ether/ethyl acetate, 3:1, v/v) furnished compound 4 as a yellow oil (1.34 g, 66.8% yield). 1H NMR (600 MHz, CD3OD) δ: 7.62 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 15.7 Hz, 1H), 7.50 (d, J = 15.7 Hz, 1H), 7.47 (d, J = 2.1 Hz, 1H), 7.25 (dd, J = 8.4, 2.1 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 2.1 Hz, 1H), 6.47 (dd, J = 8.5, 2.2 Hz, 1H), 5.29 (s, 4H), 3.91 (s, 3H), 3.81–3.73 (m, 4H), 1.22 (dt, J = 16.0, 7.1 Hz, 6H). 13C NMR (150 MHz, CD3OD) δ: 192.5, 164.8, 162.7, 150.8, 148.9, 143.0, 133.9, 131.0, 126.9, 124.8, 121.5, 117.8, 117.3, 109.0, 100.1, 95.4, 95.0, 65.6, 65.5, 56.1, 15.5, 15.4. HRMS (ESI) m/z: [M + H]+ calcd. for [C22H26O7 + H]+ 403.4511, found: 403.1760.
3.1.3 Synthesis of 4--3-methoxyphenol, 5
Compound 4 (1.2 g, 3.0 mmol) was dissolved in EtOH (40 mL) in a dried flask containing palladium on carbon (120 mg). The mixture was stirred under a hydrogen atmosphere at room temperature for 48 h, during which the reaction was monitored by TLC. Upon complete consumption of the starting material, the catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure to afford the crude product. Purification by silica gel column chromatography (petroleum ether/ethyl acetate, 5:1, v/v) furnished compound 5 as a colorless oil (1.06 g, 90.6% yield). 1H NMR (600 MHz, CD3OD) δ: 7.01 (d, J = 9.7 Hz, 1H), 6.95 (s, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 8.2 Hz, 1H), 6.38 (s, 1H), 6.29 (d, J = 8.1 Hz, 1H), 5.20 (s, 2H), 5.17 (s, 2H), 3.78–3.73 (m, 7H), 2.51 (q, J = 8.3 Hz, 4H), 1.79 (dt, J = 14.5, 7.7 Hz, 2H), 1.20 (t, J = 7.1 Hz, 6H). 13C NMR (150 MHz, CD3OD) δ: 159.7, 157.7, 148.7, 146.7, 138.8, 131.2, 123.4, 122.6, 118.9, 118.8, 107.5, 99.7, 95.6, 95.4, 65.3, 65.2, 55.6, 36.0, 33.2, 30.2, 15.5, 15.4. HRMS (ESI) m/z: [M + Na]+ calcd. for [C22H30O6 + Na]+ 413.4651, found: 413.1941.
3.1.4 Synthesis of 6ʹ,7'-dihydroxy-2-methoxy-3',4'-dihydro-2'H-spiro(cyclohexane-1,1'-naphthalene)-2,5-dien-4-one, 7
Compound 5 (59 mg, 0.15 mmol) was placed in a dry reaction flask equipped with acetonitrile (9 mL) and water (1 mL). To this solution were sequentially added potassium hydrogen persulfate (184 mg, 0.6 mmol) and tetrabutylammonium hexafluorophosphate (116 mg, 0.3 mmol). The electrocatalytic reaction was then carried out using a C-Pt electrode pair at a constant current of 8 mA at room temperature for 4 h. The progress of the reaction was monitored by TLC until the starting material was completely consumed. Upon completion, the electrolysis was stopped, and the mixture was transferred to a clean round-bottom flask, where FeBr3 (6 mg) was added and the mixture was stirred for an additional 4 h. Acetonitrile was removed under reduced pressure, and the residue was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude product. Purification by silica gel column chromatography (petroleum ether/ethyl acetate, 2:1, v/v) afforded compound 7 as a red-brown powder (30.4 mg, 74.4% yield). 1H NMR (600 MHz, CD3OD) δ: 6.94 (d, J = 9.8 Hz, 1H), 6.53 (s, 1H), 6.21 (s, 1H), 6.04 (dd, J = 9.8, 1.6 Hz, 1H), 5.70 (d, J = 1.6 Hz, 1H), 3.69 (s, 3H), 2.75–2.63 (m, 2H), 2.28–2.20 (m, 1H), 2.03–1.77 (m, 3H). 13C NMR (150 MHz, CD3OD) δ: 191.1, 184.0, 154.8, 145.8, 145.0, 130.0, 125.2, 123.7, 117.0, 114.6, 102.5, 56.5, 48.2, 34.7, 29.8, 20.6. HRMS (ESI) m/z: [M + H]+ calcd. for [C16H16O4 + H]+ 273.3079, found: 273.1120. (+)-Spirobroussonin B \([\alpha]^_\text\): + 106.18; (−)-Spirobroussonin B \([\alpha]^_\text\): − 136.00.
3.1.5 Synthesis of 4-(3-(3-(ethoxymethoxy)-4-methoxyphenyl)propyl)-3-methoxyphenol, 5a
To a solution of (E)-3-(3-(ethoxymethoxy)-4-methoxyphenyl)-1-(4-hydroxy-2-methoxyphenyl)prop-2-en-1-one (1.08 g, 3.0 mmol) in EtOH (40 mL) was added 10% palladium on carbon (120 mg). The mixture was stirred under a hydrogen atmosphere at room temperature for 48 h, and the reaction was monitored by TLC until complete consumption of the starting material. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure to give the crude product. Purification by silica gel column chromatography (petroleum ether/ethyl acetate, 5:1, v/) afforded compound 5a as a colorless oil (809.6 mg, 78.0% yield). 1H NMR (400 MHz, CD3OD) δ: 6.92 (d, J = 2.1 Hz, 1H), 6.86 (d, J = 8.1 Hz, 2H), 6.78 (dd, J = 8.2, 2.1 Hz, 1H), 6.38 (d, J = 2.4 Hz, 1H), 6.28 (dd, J = 8.1, 2.4 Hz, 1H), 5.17 (s, 2H), 3.80 – 3.72 (m, 8H), 2.51 (td, J = 7.7, 5.0 Hz, 4H), 1.85 – 1.73 (m, 2H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ: 159.6, 157.7, 149.5, 147.4, 136.9, 131.1, 123.5, 122.6, 118.9, 113.5, 107.5, 99.7, 95.5, 65.3, 56.5, 55.6, 35.9, 33.2, 30.2, 15.4.
3.1.6 Synthesis of 7'-hydroxy-2,6'-dimethoxy-3',4'-dihydro-2'H-spiro(cyclohexane-1,1'-naphthalene)-2,5-dien-4-one, 8
Compound 5a (52 mg, 0.15 mmol) was placed in a dry reaction flask equipped with acetonitrile (9 mL) and water (1 mL). To this solution were sequentially added potassium hydrogen persulfate (184 mg, 0.6 mmol) and tetrabutylammonium hexafluorophosphate (116 mg, 0.3 mmol). The electrocatalytic reaction was then carried out using a C–Pt electrode pair at a constant current of 8 mA at room temperature for 1 h. The progress of the reaction was monitored by TLC until the starting material was completely consumed. Upon completion, the electrolysis was stopped, and the mixture was transferred to a clean round-bottom flask, where FeBr3 (6 mg) was added and the mixture was stirred for an additional 4 h. Acetonitrile was removed under reduced pressure, and the residue was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude product. Purification by silica gel column chromatography (petroleum ether/ethyl acetate, 2:1, v/v) afforded compound 7 as a red-brown powder (21.6 mg, 53% yield) and compound 8 as a white solid (4.3 mg, 10% yield). 1H NMR (600 MHz, CD3OD) δ: 6.98 (d, J = 9.8 Hz, 1H), 6.69 (s, 1H), 6.24 (s, 1H), 6.08 (dd, J = 9.8, 1.6 Hz, 1H), 5.73 (d, J = 1.6 Hz, 1H), 3.83 (s, 3H), 3.72 (s, 3H), 2.79 (dt, J = 12.2, 5.6 Hz, 2H), 2.28 (ddd, J = 13.8, 11.3, 3.2 Hz, 1H), 2.04–1.83 (m, 3H). 13C NMR (150 MHz, CD3OD) δ: 191.0, 183.7, 154.5, 148.5, 146.1, 130.0, 126.6, 124.0, 114.5, 113.6, 102.6, 56.5, 56.3, 48.2, 34.7, 30.0, 20.6. HRMS (ESI) m/z: [M + H]+ calcd. for [C17H18O4 + H]+ 287.3347, found: 287.1287. (+)-Methyl spirobroussonin B \([\alpha]^_\text\): + 130.43; (−)-Methyl spirobroussonin B \([\alpha]^_\text\): −98.11.
3.1.7 Gram-scale synthesis of spirobroussonin B, via electrocatalytic oxidative coupling
Compound 5 (1.10 g, 2.82 mmol) was placed in a dry reaction flask equipped with acetonitrile (36 mL) and water (4 mL). To this solution were sequentially added potassium hydrogen persulfate (3.46 g, 11.3 mmol) and tetrabutylammonium hexafluorophosphate (2.18 g, 5.6 mmol). The electrocatalytic reaction was then carried out using a C-Pt electrode pair at a constant current of 8 mA at room temperature for 6 h. The progress of the reaction was monitored by TLC until the starting material was completely consumed. Upon completion, the electrolysis was stopped, and the mixture was transferred to a clean round-bottom flask, where FeBr3 (110 mg) was added and the mixture was stirred for an additional 4 h. Acetonitrile was removed under reduced pressure, and the residue was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude product. Purification by silica gel column chromatography (petroleum ether/ethyl acetate, 2:1, v/v) afforded compound 7 as a white amorphous powder (392 mg, 51.1% yield).





Comments (0)