4.1 Material and methods
All reagents and solvents were purchased from commercial suppliers and used without further purification. Imm-CalB lipase (lot n. 316694, 295919, 270027) was purchased by Merck (Milan, Italy) All reactions requiring anhydrous conditions were performed under a positive nitrogen flow (after vacuuming the flask), and all glassware was oven dried. Thin layer chromatography (TLC) analyses were performed using commercial silica gel 60 F254 aluminum sheets. Spots were revealed under a UV lamp (λ = 254 or 365 nm). Ninhydrin, phosphomolybdic acid, bromocresol green and Pancaldi solution-based TLC stains were used occasionally as needed. Isolation and purification of the products were performed by flash column chromatography on silica gel 60 (230–400 mesh). Optical rotations were measured using a Jasco p-2000 polarimeter. The NMR spectra were acquired using a Bruker NMR Avance NEO 400 MHz spectrometer (1H 400 MHz,13C 100 MHz). Tetramethylsilane (TMS) was used as an internal standard, and chemical shifts ( δ) are expressed in ppm. The coupling constants (J) are reported in Hz. For the NMR analysis we used CDCl3, CD3OD, D2O, DMSO-d6 as deuterated solvents. All the spectra were recorded at room temperature (298.15 K). 1H NMR signals are indicated with the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), m (multiplet). HPLC analyses were performed using a JASCO PU-980 Binary HPLC Pump, equipped with a JASCO UV-975 UV–vis detector and a JASCO LC-NetII/ADC interface box. Chiral column employed was a Kromasil 5-AmyCoat (4.6*250 mm). Injection volume: 20 μL. Flow rate: 1.0 mL min−1, n-hex: iPrOH 9:1 + 0.1% TFA (3 vs rac-3) or n-hex: iPrOH 1:9 + 0.2% formic acid. Mass spectrometry analyses were performed at the Mass Spectrometry facility of the Unitech COSPECT at the University of Milan (Italy).
4.2 Chromeazurol S (CAS) assay
CAS Reagent. 100 mL graduated glass cylinder was previously washed with 6 M HCl. A solution of hexadecyltrimethylammonium bromide (HDTMA) (22 mg in 6 mL distilled water, 10 mM) was poured into the cylinder and diluted to 40 mL with distilled water. 1.5 mL of ferric iron solution (2.7 mg of FeCl3·6 H2O in 10 mL 1 M HCl, 1 mM) was added while stirring. Finally, 7.5 mL of Chrome Azurol S (CAS) solution (9.1 mg in 7.5 mL distilled water, 2 mM) was slowly added to the mixture.
In a round bottom flask, a solution of piperazine (4.31 g in 20 mL distilled water, 2.5 M) was cooled to 0 °C and 6.25 mL of concentrated HCl was carefully added. The piperazine solution was added to the CAS/Fe3+ solution in the cylinder and then brought to a final volume of 100 mL with distilled water. Lastly, sulfosalicylic acid (102 mg) was added and dissolved to afford a blue solution, stored in a polyethylene falcon in a cool and dark place.
Chelation Assay. To prepare the stock solutions of the tested compounds, 1 mg was dissolved in 1 mL of H2O. 100 μL of CAS reagent pipetted in the well of the 96 Microwell plate, then 100 μL of the tested compound solution were added at 0.125, 0.25, 0.5 and 1 mg/mL into the same well and allowed to react for 30 min. The assay was done in triplicate. EDTA was used as a positive control, H2O as the negative control.
4.3 Chemistry4.3.1 Synthesis of triethyl 2-hydroxypropane-1,2,3-tricarboxylate (TEC)
In a two-neck round bottom flask, CA (2 g, 10.4 mmol) was dissolved in EtOH (52 mL) under nitrogen atmosphere. Thionyl chloride (2.5 mL, 34.3 mmol) was added dropwise and the mixture was heated at 78 °C under reflux for 16 h. After evaporation of the solvent, the crude mixture was dissolved in a saturated solution of NaHCO3 and the aqueous phase was extracted with ethyl acetate three times. The collected organic layers were washed with brine and dried over Na2SO4. The residue was purified by column chromatography (hexane/ethyl acetate 6:4) to afford TEC as a colorless oil (2.7 g, 94%). Rf = 0.6 (hexane/ethyl acetate 1:1 + 2 drops of acetic acid, stain: PMA). HRMS (ESI) m/z calcd for C12H20O7Na, [M + Na]+: 299.1107, found: 299.1107. 1H NMR (400 MHz, CDCl3) δ 4.31 (q, J = 7.1 Hz, 2H), 4.17 (q, J = 7.1 Hz, 4H), 2.91 (d, J = 15.5 Hz, 2H), 2.80 (d, J = 15.6 Hz, 2H) 1.33 (t, J = 7.1 Hz, 3H), 1.27 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 173.37, 169.74, 73.20, 62.28, 60.93, 43.34, 14.07, 14.01.
4.3.2 Synthesis of (R)-5-ethoxy-3-(ethoxycarbonyl)-3-hydroxy-5-oxopentanoic acid (3)
In a round bottom flask, TEC (2.7 g, 9.76 mmol) was dissolved in of phosphate buffer (pH = 8) (57 mL) and imm-CaLB (700 mg) was added. The mixture was stirred at 40 °C for 16 h. The reaction was not complete and other 200 mg of the enzyme were added and the stirring was continued for other 60 h at room temperature. The pH of the solution was adjusted to 9 with 1 N NaOH and the aqueous phase was extracted twice with diethyl ether. The aqueous phase was acidified with concentrated HCl until pH = 3 and extracted twice with ethyl acetate. The combined organic phases were washed with brine and dried over Na2SO4. After evaporation of the solvent compound 3 (2.25 g, 93%) was obtained as a yellow oil. Rf = 0.22 (DCM/MeOH 9:1, stain: Pancaldi solution). Spectroscopic data were in agreement with already reported literature [19]. [α]D = + 3.59 (c 0.2, MeOH). HRMS (ESI) m/z calcd for C10H16O7Na, [M + Na]+: 299.0796, found: 271.0794. 1H NMR (400 MHz, CDCl3) δ 4.27 (q, J = 7.1 Hz, 2H), 4.14 (q, J = 7.1 Hz, 2H), 2.91 (t, J = 15.6 Hz, 2H), 2.81 (t, J = 15.9 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H), 1.24 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 174.77, 173.19, 169.80, 73.02, 62.55, 61.11, 43.19, 43.03, 14.05, 13.99, 13.95.
4.3.3 Synthesis of 5-ethoxy-3-(ethoxycarbonyl)-3-hydroxy-5-oxopentanoic acid (rac-3)
In a round bottom flask, to a solution of triethyl citrate (100 mg, 0.36 mmol) in EtOH/H2O 1:1 (1.7 mL), 0.1 N solution of NaOH (1.8 mL) was added. The mixture was stirred at rt for 5 h, then concentrated under reduced pressure. The crude was extracted twice with ethyl acetate, the resulting aqueous phase was acidified at pH = 3 and extracted three times with ethyl acetate. The combined organic phases were washed with brine and dried over Na2SO4. After evaporation of the solvent compound 3-rac (2.25 g, 93%) was obtained as a yellow oil. Rf = 0.22 (DCM/MeOH 9:1, stain: Pancaldi solution). Spectroscopic data were in agreement with already reported literature [18]. 1H NMR (400 MHz, DMSO-d6) δ 4.10 (q, J = 7.1 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 2.84 (d, J = 15.0 Hz, 1H), 2.79 (d, J = 15.5 Hz, 1H), 2.71 (d, J = 15.0 Hz, 1H), 2.64 (d, J = 15.5 Hz, 1H), 1.19 (t, J = 7.1 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H).
4.3.4 Synthesis of tetraethyl 2,2'-((butane-1,4-diylbis(azanediyl))bis(2-oxoethane-2,1-diyl))(2R,2'R)-bis(2-hydroxysuccinate) (4) and diethyl 2,2'-((3R,3'R)-butane-1,4-diylbis(3-hydroxy-2,5-dioxopyrrolidine-1,3-diyl))diacetate (5) and diethyl (R)-2-(2-((4-((R)-3-(2-ethoxy-2-oxoethyl)-3-hydroxy-2,5-dioxopyrrolidin-1-yl)butyl)amino)-2-oxoethyl)-2-hydroxysuccinate (6)
In a two-neck round bottom flask, under nitrogen atmosphere, mono acid 3 (200 mg, 0.81 mmol) was dissolved in dry DMF (6 mL). At 0 °C TBTU (305 mg, 0.95 mmol), HOBt (128 mg, 0.95 mmol) and DIPEA (0.65 mL, 3.74 mmol) were added. After 5 min stirring, putrescine (34 μL, 0.34 mmol) was added. The reaction was left for 16 h at room temperature. After evaporation of the solvent, the crude was purified by column chromatography (DCM/MeOH 98:2 → 90:10) to obtain 5 as the main product (106 mg, 69%) as a yellow oil. Rf = 0.33 (DCM/MeOH 95:5, stain: Pancaldi solution). [α]D = –16.03 (c 0.3, MeOH). HRMS (ESI) m/z calcd for C20H28N2O10Na, [M + Na]+: 479.1642, found: 479.1644. 1H NMR (400 MHz, CD3OD) δ 4.13 (qd, J = 7.1, 1.8 Hz, 4H), 3.61–3.50 (m, 4H), 3.07 (d, J = 5.5 Hz, 2H), 3.02 (d, J = 4.0 Hz, 2H), 2.92 (d, J = 16.7 Hz, 2H), 2.66 (d, J = 18.3 Hz, 2H), 1.71–1.56 (m, 3H), 1.25 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CD3OD) δ 178.90, 175.47, 170.08, 71.77, 60.67, 41.62, 40.37, 37.65, 24.33, 13.05.
4: HRMS (ESI) m/z calcd for C24H40N2O12Na, [M + Na]+: 571.2479, found: 571.2488. 1H NMR (400 MHz, CD3OD) δ 4.24 (qd, J = 7.1, 1.6 Hz, 4H), 4.13 (q, J = 7.1 Hz, 4H), 3.21–3.15 (m, 4H), 2.95 (d, J = 15.5 Hz, 2H), 2.76 (d, J = 15.5 Hz, 2H), 2.75 (d, J = 14.6 Hz, 2H), 2.64 (d, J = 14.6 Hz, 2H), 1.30 (t, J = 7.1 Hz, 6H), 1.26 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 173.63, 170.19, 169.45, 73.72, 62.42, 61.05, 44.47, 43.13, 38.98, 29.70, 26.53, 14.09.
6: HRMS (ESI) m/z calcd for C22H34N2O11Na, [M + Na]+: 525.2060, found: 525.2059. 1H NMR (400 MHz, CD3OD) δ 4.24 (qd, J = 7.1, 1.7 Hz, 2H), 4.17–4.08 (m, 4H), 3.54 (t, J = 6.9 Hz, 2H), 3.19 (t, J = 6.8 Hz, 2H), 3.12–3.01 (m, 2H), 2.99–2.86 (m, 2H), 2.83–2.54 (m, 4H), 1.72–1.57 (m, 2H), 1.52–1.46 (m, 2H), 1.41–1.14 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 178.42, 174.58, 173.57, 170.20, 170.08, 169.33, 73.60, 72.31, 62.40, 61.32, 61.03, 44.47, 43.00, 42.07, 41.03, 38.82, 38.42, 29.67, 26.42, 24.45, 14.04.
4.3.5 Synthesis of 2,2'-((3R,3'R)-butane-1,4-diylbis(3-hydroxy-2,5-dioxopyrrolidine-1,3-diyl))diacetic acid ((R,R)-glomuferrin, 1)
In a round bottom flask, to a suspension of diethyl ester 5 (29 mg, 0.07 mmol) in distilled water (7.5 mL), imm-CaLB (230 mg) was added. The reaction was left stirring at 240 rpm at 40 °C for 16 h. The solvent was evaporated under reduced pressure to afford pure compound 1 (26.1 mg, 96%) as a colourless oil. Rf = 0.16 (DCM/MeOH 9:1, stain: Ninhydrin solution). [α]D = + 1.12 (c 0.3, DMSO). HRMS (ESI) m/z calcd for C16H19N2O10, [M – H]–: 399.1040, found: 399.1039. 1H NMR (400 MHz, D2O) δ 3.59–3.42 (m, 4H), 3.10 (d, J = 18.6 Hz, 2H), 2.99 (d, J = 16.8 Hz, 2H), 2.92 (d, J = 16.8 Hz, 2H), 2.78 (d, J = 18.6 Hz, 2H), 1.60–1.40 (m, 4H). 13C NMR (101 MHz, D2O) δ 180.40, 177.75, 173.92, 72.22, 41.56, 41.20, 38.30, 23.98.
4.3.6 Synthesis of (2R,2'R)-2,2'-((butane-1,4-diylbis(azanediyl))bis(2-oxoethane-2,1-diyl))bis(2-hydroxysuccinic acid) ((R,R)-rhizoferrin, 2)
In a round bottom flask, compound 5 (36 mg, 0.07 mmol) was dissolved in 0.2 mL of methanol, 0.6 mL of THF and 0.2 mL of water. Then LiOH monohydrate (55.13 mg, 1.31 mmol) was added. The reaction mixture was stirred for 16 h at room temperature. Other 27.56 mg of LiOH were added and the reaction mixture was stirred for further 72 h. The solvent was evaporated and the product, dissolved in 2 mL of water, was eluted through a cation exchange resin column (Amberlyst 15®). Residual water was evaporated to give product 2 (29.8 mg, quantitative yield) as a colourless glass. [α]D = – 2.61 (c 0.3, H2O). HRMS (ESI) m/z calcd for C16H23N2O12, [M–H]–: 435.1251, found: 435.1254. 1H NMR (400 MHz, D2O) δ 3.20–3.06 (m, 4H), 3.01 (d, J = 16.1 Hz, 2H), 2.75 (d, J = 16.2 Hz, 2H), 2.74 (d, J = 14.5 Hz, 2H), 2.62 (d, J = 14.5 Hz, 2H), 1.54–1.35 (m, 4H). 13C NMR (101 MHz, D2O) δ 176.62, 173.50, 170.94, 73.67, 44.63, 43.17, 38.88, 25.64.
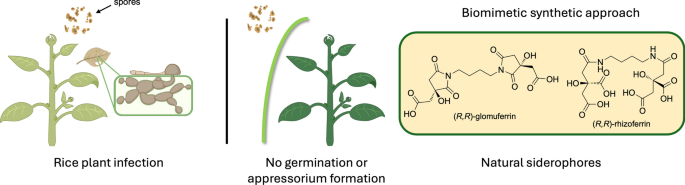
4.3.7 Synthesis of 2,2'-(butane-1,4-diylbis(3-hydroxy-2,5-dioxopyrrolidine-1,3-diyl))diacetic acid (glomuferrin, rac-1 and meso-1)
In a two-neck round bottom flask, under nitrogen atmosphere, citric acid (1 g, 5.20 mmol) was added, to a solution of putrescine (0.25 mL, 2.60 mmol) in 5 mL of xylene. The reaction mixture was stirred at 140 °C using a Dean-Stark apparatus for 24 h to remove the released water. After evaporation of the solvent the crude was purified by column chromatography (DCM/MeOH 95:5 → 80:20) to afford two major products, rac-1 as a colourless oil (12%) and meso-1 as a white solid (18%).
rac-1: HRMS (ESI) m/z calcd for C16H19N2O10, [M – H]–: 399.1040, found: 399.1035. 1H NMR (400 MHz, CD3OD) δ 3.53 (d, J = 6.4 Hz, 4H), 3.03 (d, J = 18.3 Hz, 2H), 3.02 (d, J = 16.9 Hz, 2H), 2.88 (d, J = 16.9 Hz, 2H), 2.65 (d, J = 18.2 Hz, 2H), 1.59 (t, J = 6.3 Hz, 4H). 13C NMR (101 MHz, CD3OD) δ 179.13, 175.72, 171.92, 71.73, 41.67, 40.23, 37.56, 24.21.
meso-1: HRMS (ESI) m/z calcd for C16H19N2O10, [M – H]–: 399.1040, found: 399.1036. 1H NMR (400 MHz, D2O) δ 3.66–3.53 (m, 4H), 3.15 (d, J = 18.6 Hz, 2H), 3.01 (d, J = 16.5 Hz, 2H), 2.89 (d, J = 16.4 Hz, 2H), 2.84 (d, J = 18.6 Hz, 2H), 1.65–1.58 (m, 4H). 13C NMR (101 MHz, D2O) δ 180.80, 177.99, 175.52, 72.54, 42.56, 41.76, 38.24, 23.98.
4.4 Pyricularia oryzae spore germination and appressorium inhibition
Pyricularia oryzae PO21_05 (wild-type, wt) and PO21_O7 (QoI-resistent, res) were inoculated on a complete medium (CM) [27] and incubated in the growth chamber at 24 °C in the dark for 12–14 days. Then, 2 mL of sterile water were added to the mycelium and the formed conidia were scraped from the whole mycelium surface with the help of the sterile L-shaped spatula. The spore suspension was collected and filtered through a layer of sterile gauze into an Eppendorf tube to remove mycelium. The spore concentration was estimated using the Thoma hemeacytometer and adjusted to a concentration of 2 × 104 conidia/mL.
Both glomuferrin and rhizoferrin were dissolved in water and were added to 100 μL of conidial suspension to obtain a final concentration of 5–2-0.5–0.5 mM. Conidia suspended in water were considered control. Azoxystrobin (Amistar, Syngenta) was used as a reference compound of fungal germination inhibition at concentrations 100 and 10 µM a.i. Twenty μL of the conidial suspension in three replicates were applied on a microscopic cover slide and incubated in a wet chamber for 24 h at 24 °C in the dark. The germination of 100 randomly chosen conidia for each treatment and replica was determined. The conidia were assigned into germination classes: NG = not germinated, G = germinated, and A = germinated with appressorium.
To assess the activity of the compounds in iron homeostasis, their activity was assessed in the presence or absence of Fe3+ ions (5 µM FeCl3), as previously described [5]. To the spore suspensions, FeCl3 (5 μM) was added at 0 h post inoculation (hpi), followed by the treatment with 2 mM glomuferrin or rhizoferrin at 4 hpi in three replicates. The conidia were incubated and assessed as described above.
4.5 Statistical analysis
The statistical analyses were performed using R software, version 4.4.0, [28] within the RStudio interface, version 2025.9.1.401 [29]. The percentage data of the spore germination and appressorium formation were arcsine transformed and submitted to ANOVA, followed by Tukey’s post hoc test for multiple comparison (p = 0.05), using the TukeyC package [30].





Comments (0)