
Remember me
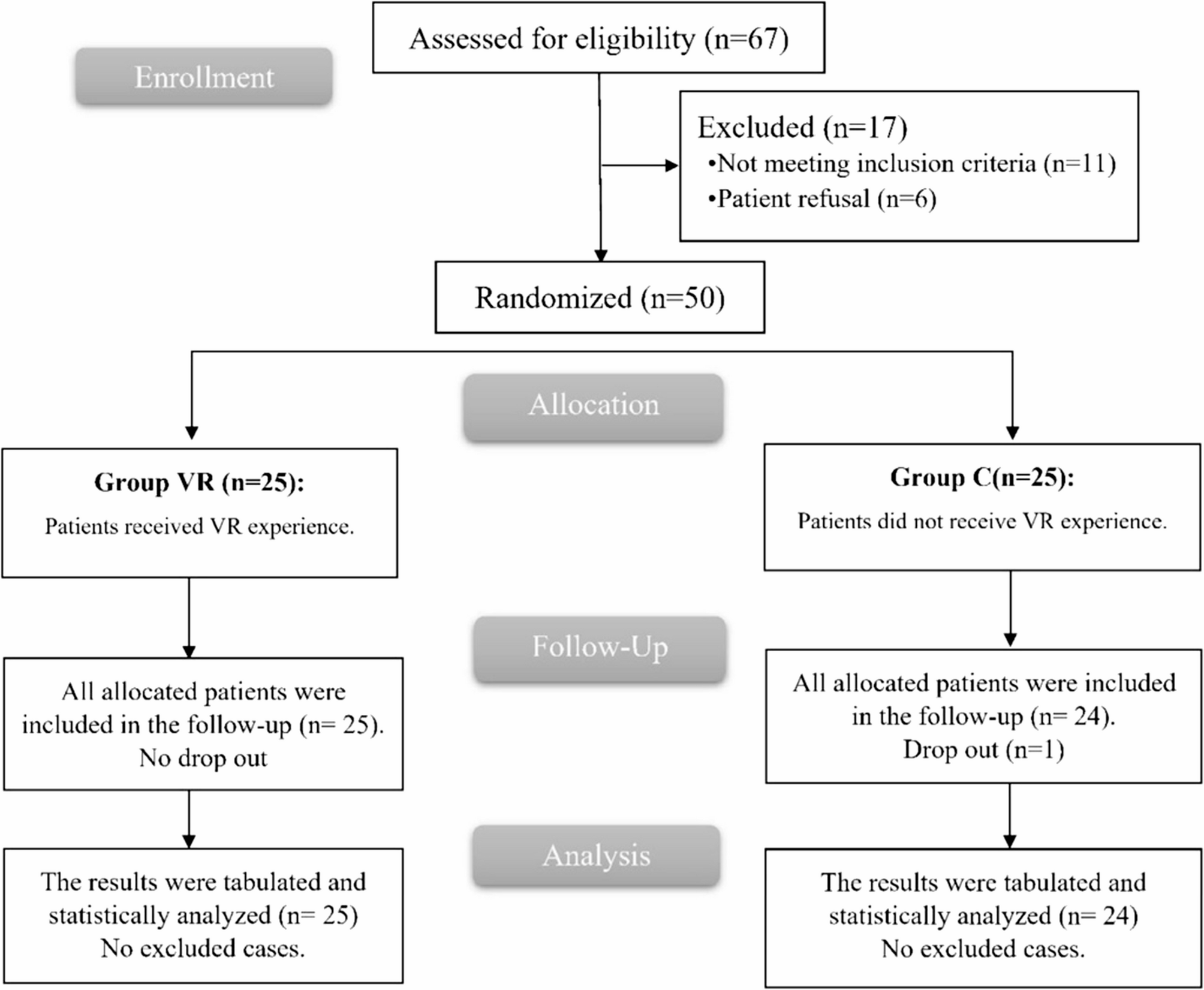
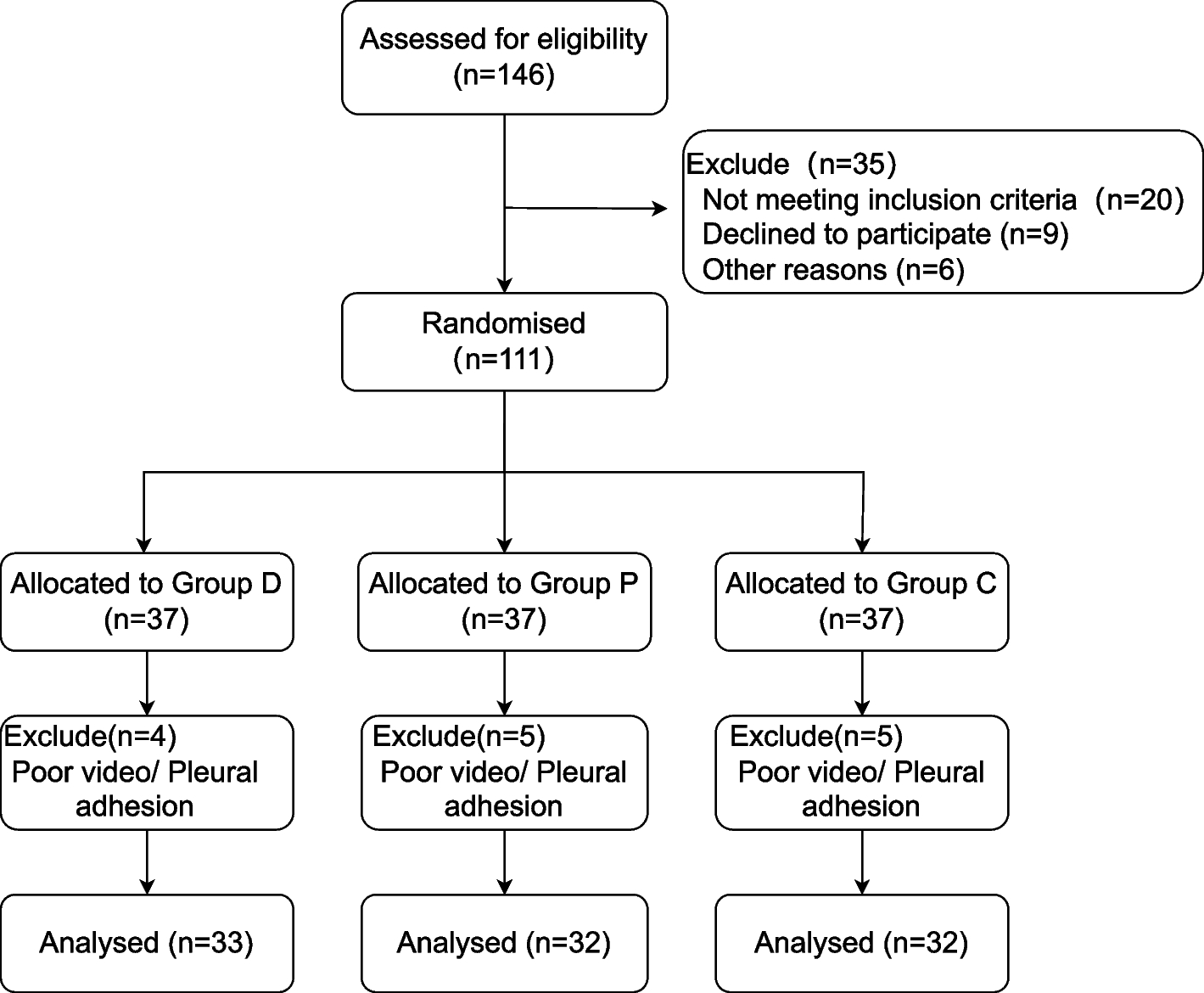
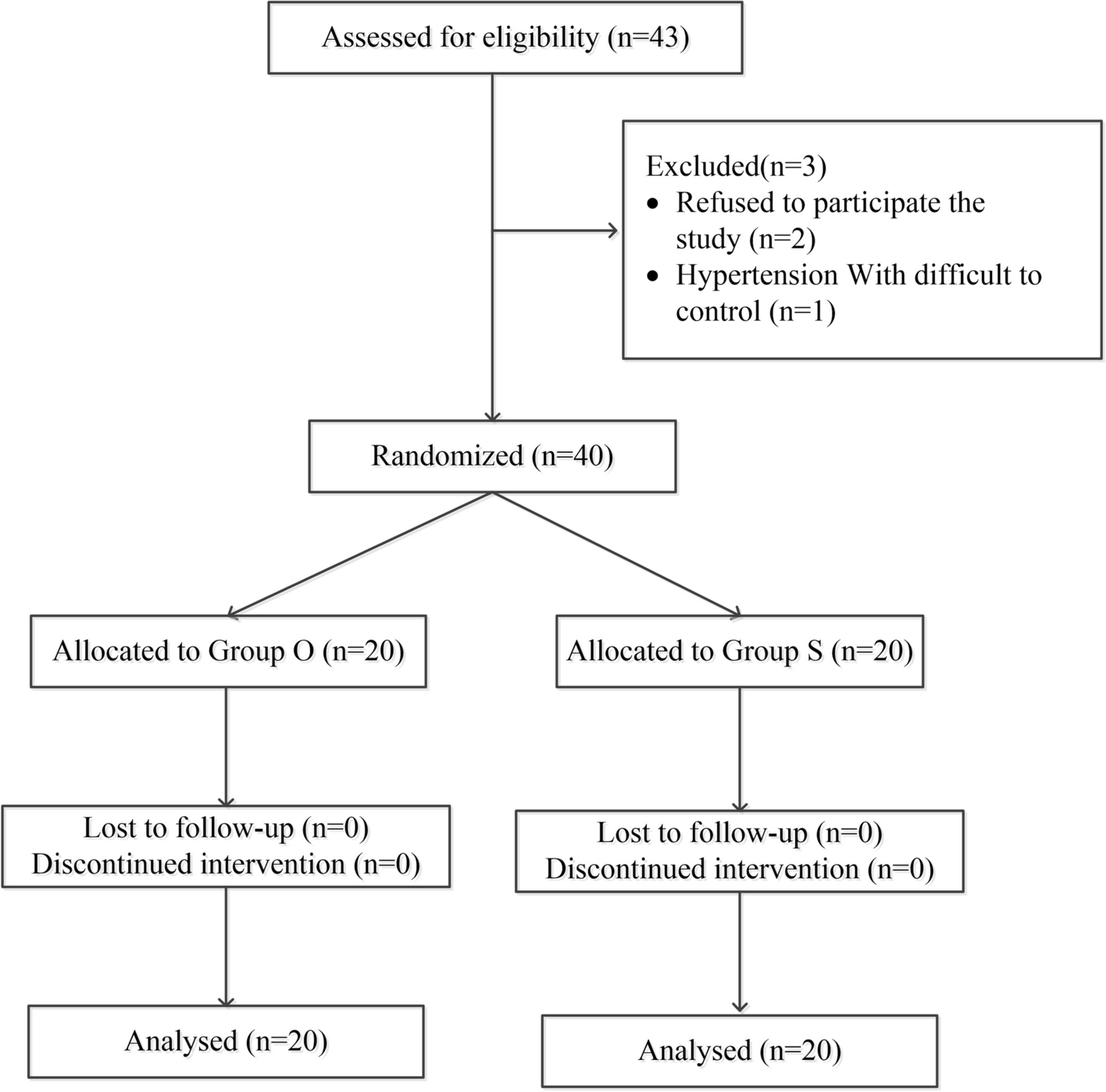
This prospective, double-blind, randomized controlled trial was approved by the Human Research Ethics Committee of the Third Affiliated Hospital of Anhui Medical University on March 28, 2022 (Approval No.: 2022–021-01, Principal Investigator: Professor Luo). It has been registered in the China Clinical Trial Registry (registration number: ChiCTR2200066896, Principal Investigator: Hong Luo, registration date: December 21, 2022). The protocol was explained to patients and written informed consent was obtained from all participants before starting the trial. All procedures were conducted following relevant guidelines and regulations. We followed the Consolidated Standards for Reporting Trials (CONSORT) reporting guidelines (Fig. 1).
Fig. 1
CONSORT patient enrolment diagram. CONSORT, Consolidated Standards of Reporting Trials
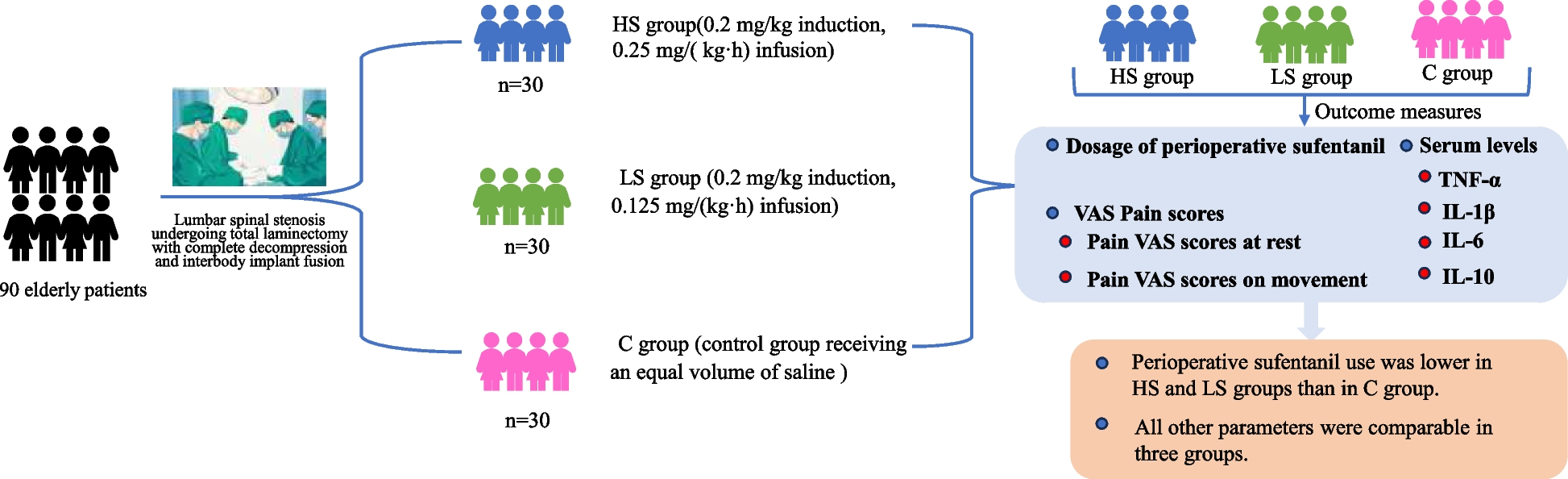
ParticipantsThe trial enrolled patients aged 65 to 89 years with an American Society of Anesthesiologists (ASA) physical status classification of II to III (I for healthy patients, II for patients with mild systemic disease, III for patients with severe systemic disease, and IV for those with severe systemic disease with a stage of loss of function). We included elderly patients with lumbar spinal stenosis who visited the Department of Spine and Orthopaedics of the Third Affiliated Hospital of Anhui Medical University between December 2022 and February 2024 and were treated with total laminectomy and complete decompression interbody implant fusion under general anesthesia. All participants were included after signing a written informed consent form.
The inclusion criteria were as follows: (1) age between 65 and 89 years; (2) imaging showing definite manifestations of lumbar spinal stenosis; and (3) patients successfully completed surgery.
The exclusion criteria were as follows: (1) patients refused to participate in this study; (2) hypersensitivity or contraindication for esketamine or other drugs routinely used intraoperatively; (3) patients with significant preoperative neuropsychiatric and cognitive disorders; (4) those with severe respiratory-circulatory disorders; (5) those with severe hepatic or renal dysfunction; (6) patients with severe and poorly controlled hypertension (systolic blood pressure > 180 mmHg and/or diastolic blood pressure ≥ 110 mmHg).
Randomization and blindingDuring the preoperative visits, the investigators obtained detailed information about patients' physical condition and basic information (including gender, age, height, weight, and years of schooling, etc.).The experimental protocol was explained to the patients and their families, and written informed consent was signed by the patients and their legal guardians. One day before surgery (POD-1), two researchers collected 3 ml of venous blood from the patients (collected at 6–7 am). The patients were assessed for preoperative pain using pain visual analog scale (VAS). SPSS 26.0 software (IBM SPSS, Armonk, NY, USA) randomly assigned all participants in a ratio of 1:1:1 into the esketamine group (HS group), esketamine (LS group), and control group (C group). Nurse anesthetists completed the randomization and treatment assignment the morning before surgery. Nurses prepared the drugs by grouping and calculated the dose of esketamine (2 ml: 50 mg, Hengrui Induction, Jiangsu, China) for anesthesia induction and maintenance based on patients' body weight. Esketamine was diluted to 5 ml in saline for induction and diluted to 20 ml in saline for maintenance. It was kept in opaque, sterile anesthetic syringe Tray, and was then given to the hands of the anesthesiologist on duty that day. Patients, anesthesiologists, nurse anesthetists, and investigators responsible for patient recruitment, data collection, and follow-up assessment were blinded to clusters.
Anesthesia and perioperative analgesia managementThe patients were routinely fasted and abstained from food and drink for 8 h preoperatively. After entering the operation room, a peripheral vein was opened and infused with lactated Ringer's solution. Electrocardiogram, pulse oximetry, invasive blood pressure (BP), carbon dioxide waveform, inhalation anesthetic concentration, electroencephalographic bifrequency index (BIS), nasopharyngeal temperature, and urine output were intraoperatively monitored.
The HS and LS groups received esketamine (0.2 mg/kg) as induction medication, whereas saline was used in group C. Subsequently high-dose esketamine (0.25 mg/(kg-h)) was used as the maintenance drug in the HS group, low-dose esketamine (0.125 mg/(kg-h)) was used as the maintenance drug in the LS group, and an equal volume of saline was used to maintain anesthesia in the C group. Other procedures were the same in the three groups.
Anesthesia was induced by intravenous infusion of penehyclidine Hydrochloride Injection (0.2 mg), dexamethasone (10 mg), midazolam (0.03 mg/kg), etomidate (0.2 mg/kg), sufentanil (0.3 μg/kg), and cis-atracurium (0.2 mg/kg). Tracheal intubation was performed under visual laryngoscopy when the patient was unconscious and the jaw was relaxed. Mechanical ventilation was done after intubation. End-expiratory CO2 pressure (PetCO 2) was maintained between 35 and 45 mmHg by adjusting tidal volume (6–8 mL/kg) and respiratory rate (12–14 breaths/min). Subsequently, remifentanil (6–18 µg/(kg-h)) and propofol (3-6 mg/(kg-h)) were infused to maintain anesthesia, BIS was maintained at 40–60, and sufentanil was infused intermittently at 5–10 ug/dose based on the BIS value. The indexes of BP and HR parameters, and the BP and HR were maintained at ± 20% of the basal value. If they exceeded this threshold, vasoactive drugs were given when needed. Cisatracurium (1–2 mg) was pushed intermittently as needed for the procedure. Esketamine and saline were discontinued at the time of surgical suturing. Propofol and remifentanil were discontinued at the end of the procedure. After the patient regained consciousness and confirmed that the patient responded rapidly to verbal commands, the airway protective reflexes recovered well, the voluntary tidal volume was > 6 ml/kg, and the respiratory rate was smooth and regular. The endotracheal tube was withdrawn, and the patient was transferred to the PACU for monitoring. If the visual analog scale (VAS) of pain was ≥ 4 points, sufentanil was given (0.1 µg/kg), and if the pain was not relieved for 30 min, sufentanil was given at 0.05 µg/kg. When the patients had the modified Aldrete score (with a total score of 0–10. A score of 10 indicated the best possible clinical condition) ≥ 9 [13], they were returned to the general ward. All patients were connected to an patient controlled intravenous analgesia (PCIA) device for postoperative in-ward analgesia, which was configured using 100 µg sufentanil + 100 µg dexmedetomidine + 20 mg metoclopramide hydrochloride + saline in a total volume of 150 mL, with no background dosage, a single dose of 2 mL, and a lockout time of 15 min.
Intraoperative hypotension was defined as a decrease in MAP of more than 20% from the preoperative level, and intraoperative hypertension was defined as an increase in MAP of more than 20% from the preoperative level or a systolic blood pressure of > 180 mm Hg. Dobutamine 2 mg/dose was given in case of intraoperative hypotension, and uradil 10 mg/dose was given in case of hypertension. Atropine 0.3 mg/dose/minute was applied for heart rate less than 50 beats, and esmolol 10 mg/dose/minute was applied for a heart rate more than 100 beats.
Outcome measuresThe primary outcome was the dose of sufentanil used in the perioperative period (cumulative PCIA dosage from the induction of anesthesia to 72 h postoperatively). Patient pressed the intravenous self-contained analgesia device on demand with a single dose of 2 ml). Secondary outcomes were as follows: the concentrations of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-10 (IL-10) serum biomarker at 1 day preoperatively (POD-1), postoperative day 1 (POD1), postoperative day 3 (POD3), and 7 days postoperatively (POD7) (POD refers to the perioperative period), mean arterial pressure and heart rate of each group at each time point before anesthesia (T0), immediately after intubation (T1), 5 min after intubation (T2), at the time of surgical incision (T3), at the time of extubation (T4), and 30 min after surgery (T5), frequency of intraoperative vasoactive drug use, the visual analog scale (VAS) of pain at rest and during exercise (with a score of 0 indicating no pain and a score of 10 indicating severe pain) [14] on POD-1, POD1, POD3, and POD7, intraoperative bleeding, urine output, fluid replacement volume, operative time, anesthesia time, awaking time, PACU stay,extubation time, propofol and remifentanil dosage, postoperative hospital stay, and the incidence of adverse events within 5 days after surgery (respiratory depression (pulse oximetry < 90%), nausea, vomiting, and psychiatric adverse effects (hallucinations, nightmares, diplopia, somnolence, dizziness, etc.).
Measurement of serum biomarkers: 3 ml of venous blood was collected, and centrifuged at 1000 rpm for 10 min within 15 min. The supernatant was collected and stored at -80 °C. Serum biomarkers were measured using ELISA. The concentrations of TNF-α, IL-1β, IL-6, and IL-10 in the peripheral blood were measured by ELISA. All steps were conducted strictly following the instructions of the ELISA kit (Shanghai Jianglai Biotechnology Co., Ltd., Shanghai, China).
Sample size calculationIn this study, the sample size was calculated using perioperative sufentanil use as the main indicator. The sample size of all three groups was designed as 1:1:1, and SPSS 26.0 software was utilized for sample size calculation. Based on the results of the pilot study, the mean sufentanil dosage was 60.42 µg, 65.67 µg, and 70.28 µg in groups HS, LS, and C, respectively, and the standard deviation was set to ± 9.54. We considered ß = 0.1 and α = 0.05 for power analysis. In addition, we considered 10% shedding, exclusion factors and the need for safety observations, it was estimated that 84 patients needed to be included and 97 patients were finally included.
Statistical analysisStatistical analysis was conducted using SPSS 26.0 software. The normal distribution of continuous variables was evaluated using the Kolmogorov–Smirnov test. Continuous data are expressed as mean (SD) and compared using one-way ANOVA. Non-normally distributed data were analyzed using the Kruskal–Wallis test and reported as median (IQR). Categorical variables are expressed as the number of patients (n%) and were processed using the chi-square test or Fisher exact probability test. Comparisons between multiple groups were conducted using one-way ANOVA plus two-way LSD-t test. Repeated observations were analyzed using repeated measures ANOVA. Two-by-two intergroup comparisons were conducted using LSD-t test, and two-by-two time comparisons were conducted using t-test. Two-sided α = 0.05 was considered for interpreting differences. Repeated-measures analysis and split test for multiple comparisons were adjusted to the test level following the Bonferroni correction method. p-value < 0.05 was considered statistically significant.
Comments (0)