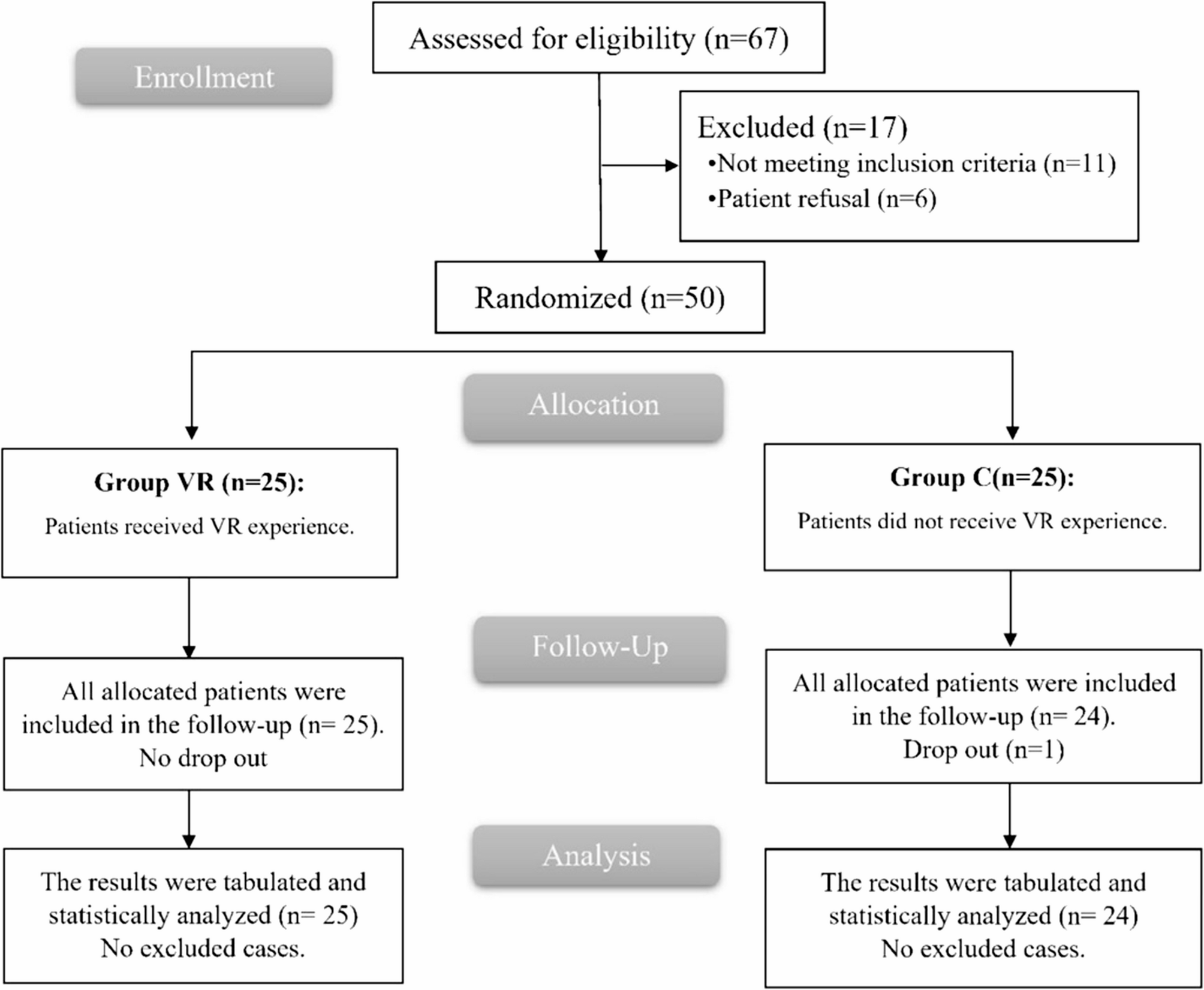
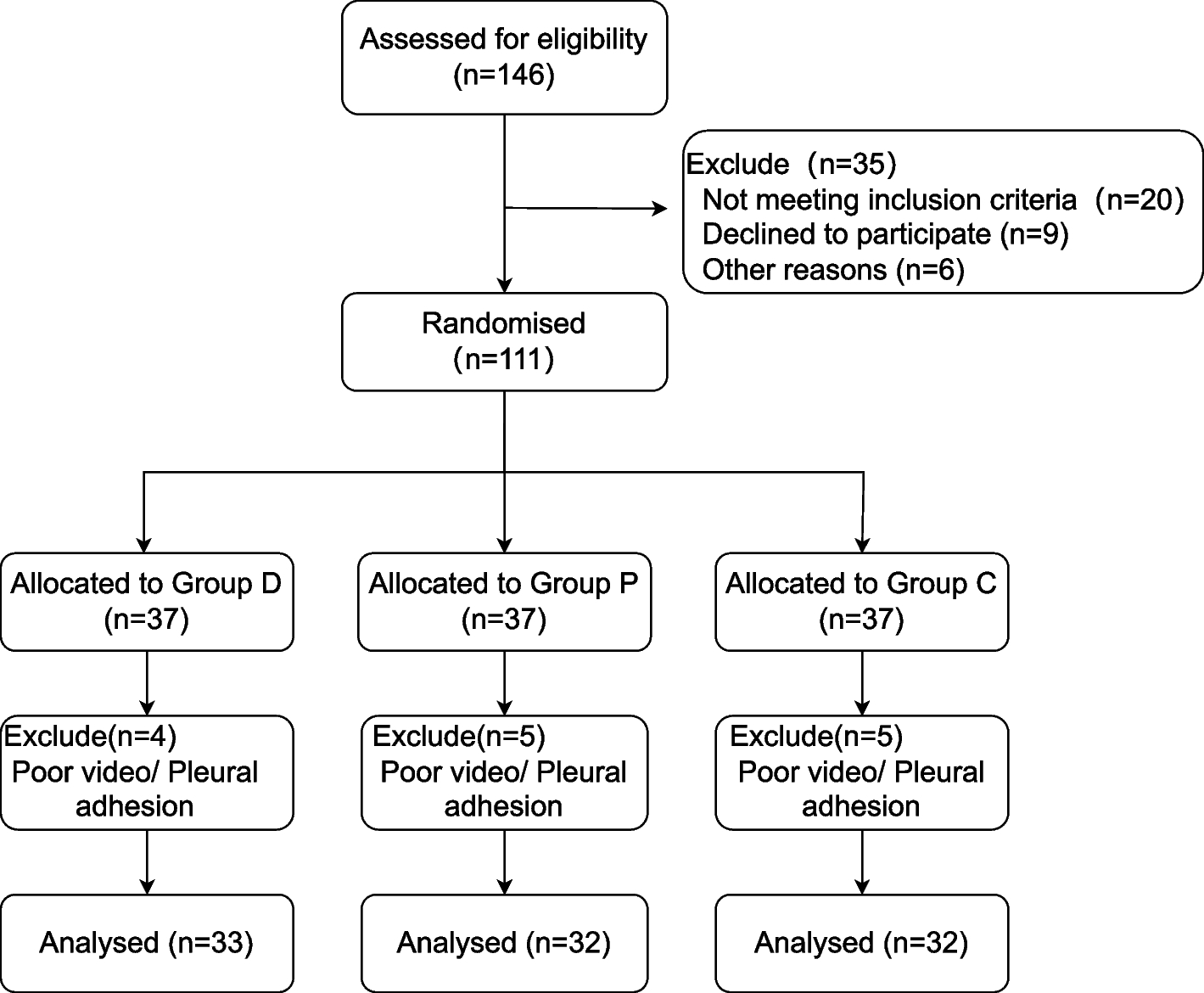
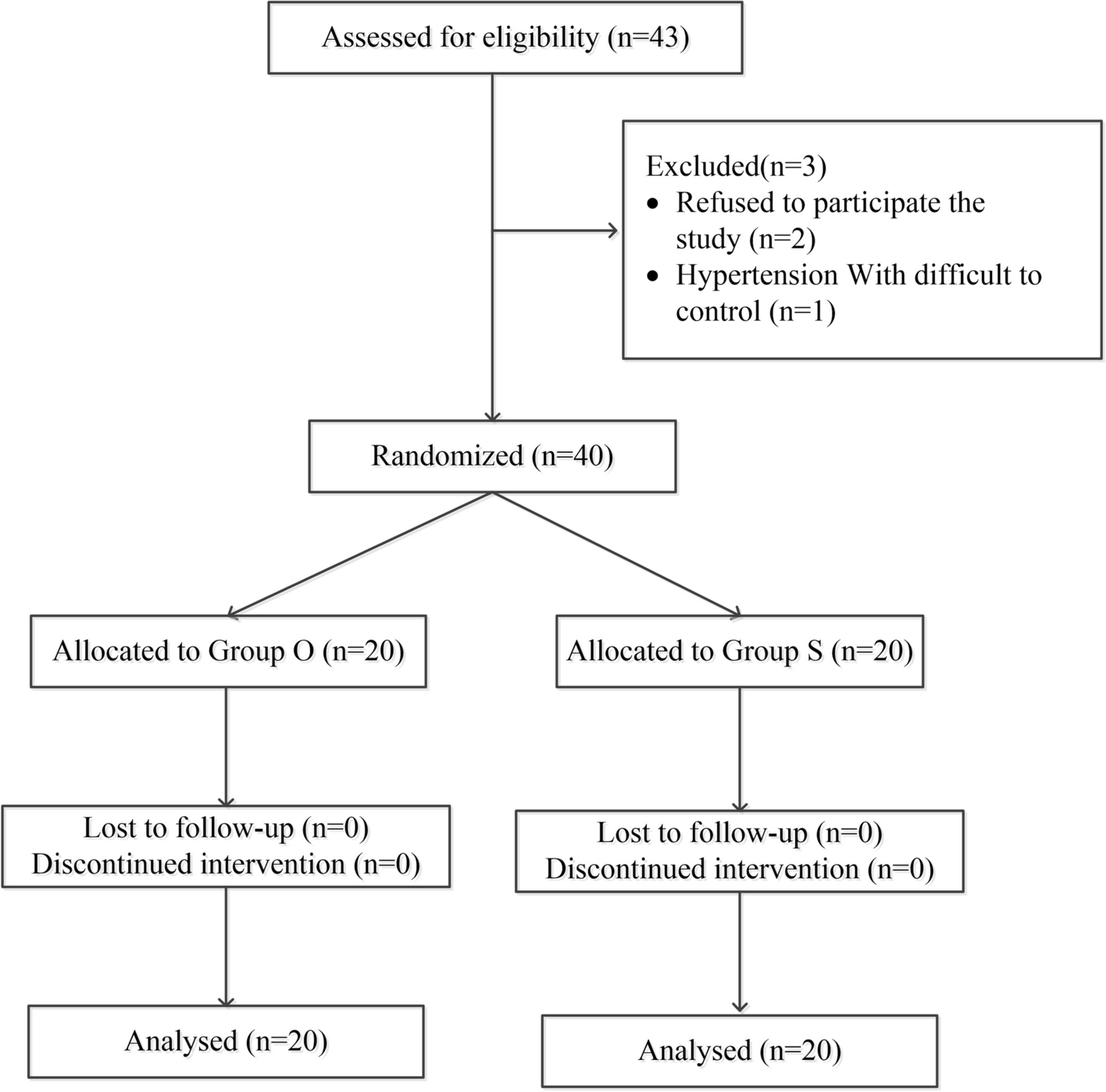
This open-label, controlled trial with random design was conducted on 50 participants of age 21 or older, both sexes, with a physical status of I-III as defined by the American Society of Anesthesiologists (ASA), listed for elective THA performed under spinal anesthesia (SA) from 19th October 2023 till 5th August 2024.
This research was approved by the Institutional Ethical Committee, Faculty of Medicine, Tanta University, Tanta, Egypt, (Approval code: 36264PR331\9\23). The trial was registered https://clinicaltrials.gov/study/NCT06088069?id=NCT06088069&rank=1 (ID: NCT06088069), Principal investigator: (SAAD AHMED MOHARAM, Date of registration: 18-10-2023). This study was conducted in compliance with the Helsinki Declaration. All subjects who participated in the trial given written, informed consent. Prior to patient enrollment.
Patients were excluded if they had adrenal insufficiency, blindness, deafness, cerebrovascular disease, chronic sedative or narcotic use, substance addiction, claustrophobia, psychiatric-cognitive dysfunction, and uncooperative behavior.
Randomization
The patients were allocated randomly using computer-generated randomization numbers parallelly to two equal groups: Group VR and Group C (control group), which did not receive any VR experience. Due to the difference in technique, the study was conducted in an open-label manner.
Preoperatively, a detailed history was taken, a clinical examination was performed, and routine laboratory investigations were conducted.
All patients underwent spinal anesthesia performed under strict aseptic conditions. A 25G Quincke spinal needle was inserted at the L3–L4 interspace, and 2.5 mL of 0.5% hyperbaric bupivacaine was administered intrathecally. No intrathecal adjuvants (such as fentanyl or morphine) were used to ensure standardization of the anesthetic technique across the study groups.
The State Anxiety Inventory (STAI-S) for anxiety was demonstrated to patients, the Perceived Stress Scale (PSS-10) for stress, and the Numerical Rating Scale (NRS) for pain. The paramedical team cordially welcomed and made necessary arrangements for patients participating in the trial to guarantee a seamless ambulant medical encounter.
In the VR group, patients were enveloped in a serene environment with nature scenes and audio features of soft music, completely isolating them from the actual world for a period of 15 min before surgery and during the procedure using VR glasses with an audio headset.
Heart rate (HR) and mean arterial blood pressure (MAP) assessed at baseline, before SA, and at 5, 10, 15, 30, 60, 90, and 120 min during the surgery, as well as at the end of surgery.
The Arabic-validated versions of the STAI-S [20] and PSS-10 [21] were measured at three different stages: 15 min before the surgery (baseline), before SA, and immediately postoperative.
Intraoperatively, SA was carried out under complete aseptic conditions. If the patient showed signs of anxiety, haloperidol was administered (2.5 mg at gradually increasing doses until the target effect was reached), and total haloperidol consumption was documented.
Postoperatively, the NRS was recorded 30 min, 2, 4, 6, 12, 18, and 24 h following the operation. All patients were given routine analgesia as 1 g of paracetamol IV. If their NRS score exceeded three, they received rescue analgesia with IV pethidine (0.5 mg/kg). The total amount of pethidine consumed within the first 24 h post-surgery and the time to the first dose of rescue analgesia were documented.
The levels of serum cortisol were assessed before and six hours following the surgical procedure. The level of patient satisfaction was evaluated using a 3-point scale (1 = unsatisfied, 2 = neutral, 3 = satisfied) after discharge to the ward.
Anxiety was the primary outcome of the research. Included in the secondary outcomes were hemodynamic parameters, stress, pain score, the cumulative opioid dose administered within the initial twenty-four hours following the operation, total haloperidol dose during the operation, serum cortisol, and patient satisfaction.
Sample size calculation
Sample size calculations were conducted using G*Power 3.1.9.2 (Universitat Kiel, Germany). A pilot study that was not published was conducted, with each category contains five cases., and we found that the mean (± standard deviation (SD)) of STAI score before SA (the primary outcome) was 36.2 ± 7.8 in group VR and 44.2 ± 9.67 in group C. The total number of samples was determined by the following: effect size of 0.906, the study’s power is 80%, and the confidence level is 95%, the group ratio is 1:1, and The inclusion of four cases in each cohort was necessary to overcome drop-out. Consequently, we recruited 25 patients for each group.
Statistical analysis
Utilizing SPSS v27 (IBM©, Armonk, NY, USA), statistical analysis was carried out. To check if the data was distributed normally, we used histograms and the Shapiro-Wilks test. The unpaired Student’s t-test was employed to evaluate the quantitative parametric data, which were presented as mean and SD. The median and interquartile range (IQR) were used to assess quantitative non-parametric data, which were expressed as the Mann-Whitney test method. The qualitative variables, which were expressed as frequency and percentage, were analyzed using either the Chi-square test or Fisher’s exact test, as appropriate. A two-tailed P value that was less than or equal 0.05 was considered statistically significant.

Comments (0)