Study Design
This study was designed as a retrospective cohort study conducted at the Affiliated Hospital of Qingdao University. The study was conducted retrospectively from October 2024 to November 2024 at the Affiliated Hospital of Qingdao University. Participants’ clinical data were reviewed and analyzed on the basis of their treatment with chiglitazar, semaglutide, or pioglitazone during this period. General patient data, such as height, weight, and age, were collected by trained medical staff and kept strictly confidential. The collected information was used solely for the purposes of this study and was not disclosed or repurposed without explicit authorization.
Participants were screened according to pre-established inclusion and exclusion criteria and assigned to the chiglitazar, semaglutide, or pioglitazone groups on the basis of their prescribed medications. Each group underwent a 4-week treatment regimen as follows: oral administration of 20 mg of chiglitazar daily in the chiglitazar group, subcutaneous injection of 0.25 mg of semaglutide weekly in the semaglutide group, and oral administration of 30 mg of pioglitazone daily in the pioglitazone group. Data collection: (1) before treatment—blood glucose monitoring, all participants underwent 3–5 days of continuous five-point blood glucose monitoring, including fasting plasma glucose (FPG) and nocturnal glucose fluctuations. Sample collection—baseline serum and urine samples were collected to evaluate metabolic and biochemical indicators, such as lipid metabolism, liver function, and renal function. (2) After treatment—blood glucose monitoring, at the end of the treatment period, five-point blood glucose monitoring was repeated over 3 to 5 days to record fasting plasma glucose (FPG), the lowest nocturnal glucose levels, and glucose fluctuation patterns. Sample collection—post-treatment serum and urine samples were collected to compare changes in metabolic and biochemical indicators before and after treatment. (3) Adverse event recording: adverse events during the treatment period were meticulously recorded, including their frequency, type, and severity. These data were compared among groups to evaluate the safety and tolerability of each medication.
Ethical Approval
This study was conducted in accordance with ethical standards, including approval from the Medical Ethics Committee of the Affiliated Hospital of Qingdao University (ethics approval no. QYFY WZLL 29564). Written informed consent was obtained from all participants, and data confidentiality was strictly maintained throughout the study. No participants’ personal information was disclosed or repurposed for nonresearch purposes. The authors declare no conflicts of interest regarding this study. All medications utilized in this research (chiglitazar, semaglutide, and pioglitazone) were prescribed on the basis of clinical judgment and were not provided by pharmaceutical companies for research purposes. The study was independently designed and executed without direct funding or influence from external commercial entities.
Data Analysis
The efficacy of each medication was assessed by comparing the changes in key indicators before and after treatment within each group. These indicators included insulin resistance, lipid metabolism disorders, liver function abnormalities, and renal function impairment. Differences among the three groups were also analyzed to evaluate the relative effectiveness of the medications. Additionally, adverse events were incorporated into a safety analysis to provide a comprehensive evaluation of the clinical utility of the medications. This integrated assessment aimed to determine the overall value of the drugs in clinical practice.
Patient PopulationInclusion Criteria
1.
Age between 15 and 85 years.
2.
Body mass index (BMI) ranging from 18.5 to 40 kg/m2.
3.
Poor glycemic control, with hemoglobin A1c (HbA1c) levels remaining between 7.5 and 10% despite strict adherence to dietary and exercise interventions.
4.
No prior use of GLP-1 receptor agonists or PPAR agonist medications.
5.
No prior exposure to chiglitazar, semaglutide, or pioglitazone.
Exclusion Criteria
1.
History of acute diabetic complications, including diabetic ketoacidosis or hyperosmolar hyperglycemic syndrome.
2.
Presence of refractory hypertension requiring four or more medications for control.
3.
Triglyceride (TG) levels > 500 mg/dL.
4.
Current treatment with fibrates or other medications that alter glucose levels (including corticosteroids, steroids, and receptor blockers).
5.
History of transient ischemic attack, cerebrovascular accident, or unstable angina within 6 months prior to screening.
6.
Coexisting pancreatic, hepatic, or renal conditions, such as cirrhosis, active hepatitis, or impaired renal function (eGFR < 60 mL/min/1.73m2).
7.
Diagnosis of malignancy or other severe illnesses within 5 years prior to screening.
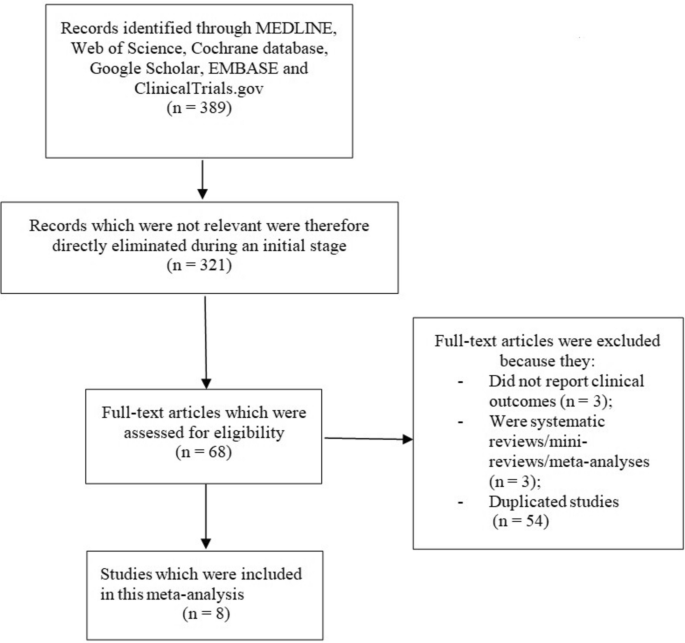
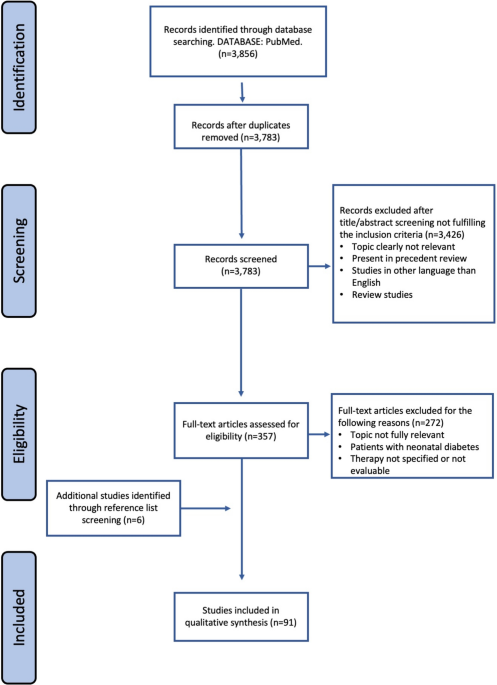
In this retrospective study, we initially reviewed the medical records of 200 candidates who were being treated for type 2 diabetes (T2D) at the Affiliated Hospital of Qingdao University. After screening, 175 patients were eligible and included in the study. However, 16 patients discontinued treatment before completing the 4-week regimen. These patients were excluded from the final analysis because they refused to undergo follow-up blood tests after the 4-week treatment period, and therefore, their post-treatment data could not be collected. It is important to note that these 16 patients were initially eligible according to the inclusion criteria, and their exclusion was solely due to the lack of follow-up data. They were not excluded because of failure to meet the inclusion criteria.
Study Endpoints and Assessments
All participants underwent comprehensive metabolic and biochemical evaluations prior to initiating treatment. The assessments included the following parameters: triglycerides (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), free fatty acids (FFA), serum uric acid (UA), alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen, serum creatinine, estimated glomerular filtration rate (eGFR), total bilirubin, albumin, urinary albumin-to-creatinine ratio (UACR), fasting insulin, and fasting C-peptide levels. After completing the 4-week treatment period, the key indicators (TG, TC, LDL-C, HDL-C, FFA, UA, ALT, AST, and UACR) were reevaluated to assess changes and treatment outcomes.
On the basis of published methodologies[7,8,9,10,11,12], the following metabolism-related indices were calculated: Metabolic Score for Insulin Resistance (METS-IR), Uric Acid to HDL Ratio (UHR), Zhejiang University Fatty Liver Index (ZJU), LTI (Liver Metabolic Index), and Atherogenic Index of Plasma (AIP). These indices provided a comprehensive quantification of peripheral insulin sensitivity, systemic inflammatory response, liver metabolic function, coronary atherosclerosis risk, and the overall level of T2D-related metabolic disorders.
Overview of Dawn Phenomenon Intensity
According to relevant studies, the intensity of the dawn phenomenon (DP) is quantified as the difference between fasting blood glucose (FBG) upon waking and the lowest nocturnal blood glucose level, typically measured at 3:00 AM [13]. Following the initiation of treatment and enrollment in the study, blood glucose levels at 3:00 AM and FBG were measured continuously over 3 to 5 days using a fingertip blood glucose monitor.After 4 weeks of treatment, the same method was employed to monitor blood glucose levels at 3:00 AM and FBG over 3 consecutive days. To minimize analytical errors, measurements taken during hypoglycemic episodes (blood glucose < 70 mg/dL) were excluded from the analysis. For each participant, the average values of DP intensity, 3:00 AM blood glucose, and FBG were calculated for analysis.
During the study period, all participants received standardized dietary and exercise guidance, with instructions to consume three meals at consistent times each day. The frequency and characteristics of hypoglycemic events (blood glucose levels < 70 mg/dL) or other abnormalities were meticulously recorded for each participant.
Statistical Analysis
All statistical analyses were performed using SPSS Statistics version 29 (IBM Corp., Armonk, NY, USA). The normality of continuous variables was assessed using the Shapiro–Wilk test. Data following a normal distribution were expressed as mean ± standard error of the mean (SEM), while non-normally distributed data were represented as median (interquartile range, IQR). Categorical variables were summarized as counts and percentages.
Baseline Characteristic Comparison
The baseline characteristics of the three patient groups (chiglitazar, semaglutide, and pioglitazone) were compared as follows: for continuous variables with a normal distribution, one-way analysis of variance (ANOVA) was used. For non-normally distributed variables, the Kruskal–Wallis test was employed. For categorical variables, the chi-square test (χ2) was applied. Changes in key metabolic parameters before and after treatment within each group were analyzed using paired t-tests for normally distributed data or Wilcoxon signed-rank tests for non-normally distributed data. The key metabolic parameters analyzed included fasting plasma glucose (FPG), triglycerides (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), free fatty acids (FFA), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and urinary albumin-to-creatinine ratio (UACR). To compare the post-treatment changes in metabolic and biochemical parameters among the three groups, ANOVA or Kruskal–Wallis tests were used. Post hoc pairwise comparisons with Bonferroni correction were performed to identify significant differences between groups.
Specific Indicator Analysis
(1) Insulin resistance and metabolic indicators: changes in METS-IR scores before and after treatment were analyzed to evaluate improvements in peripheral insulin sensitivity. (2) Lipid metabolism: the lipid profile, including triglycerides (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C), as well as related indices, such as the Lipid Triad Index (LTI), were examined to quantify the correction of lipid metabolism disorders. (3) Liver and renal function: changes in alanine aminotransferase (ALT), aspartate aminotransferase (AST), and the Zhejiang University Index (ZJU) were analyzed to assess improvements in liver function and hepatic lipid levels. The urinary albumin-to-creatinine ratio (UACR) was evaluated to determine the renal protective effects of the medications. (4) Cardiovascular risk: changes in the Atherogenic Index of Plasma (AIP) and LTI were analyzed to assess the risk of atherosclerosis. (5) Dawn phenomenon analysis: the intensity of the dawn phenomenon was quantified as the difference between fasting plasma glucose (FPG) and the lowest nocturnal blood glucose level. The mean FPG, nocturnal blood glucose levels, and dawn phenomenon intensity were calculated for each group before and after treatment. Intra-group and inter-group comparisons were conducted to evaluate the treatment effects on glucose variability and the dawn phenomenon.
Correlation Analysis
Dawn phenomenon intensity and key metabolic parameters: the relationship between changes in dawn phenomenon intensity and key metabolic parameters, including METS-IR, lipid metabolism indices, and the inflammatory marker UHR, was assessed using Pearson or Spearman correlation analysis, depending on the data distribution. Liver function and renal function indicators: the correlation between liver function indicators (ALT, AST, and ZJU) and renal function indicators (UACR and eGFR) was analyzed to explore potential interdependencies and shared mechanisms.
Result Analysis
Significance levels were indicated as p < 0.05 or p < 0.01, with all statistical tests conducted as two-tailed tests. A significance threshold of 5% was set for all analyses.

Comments (0)