
Remember me
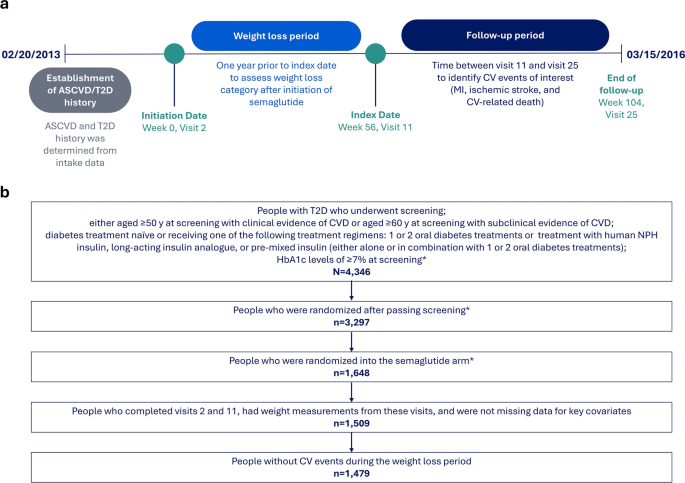
The Dia-NeP study is a multicenter, prospective, exploratory study to be conducted in Japan between March 2025 and August 2026 in patients with T2DM (Fig. 1 and Table 1). The study investigators and their institutions are presented in Supplementary Table 1. The study consists of two parts, a baseline survey and an interventional study. The baseline survey is the observational part designed to investigate the prevalence of DPN and DPNP in patients with T2DM and their background factors; the interventional study is a single-arm, open-label study designed to examine the efficacy and safety of mirogabalin in patients with DPNP.
Fig. 1
Study design. BID twice daily, CrCL creatinine clearance, DPNP diabetic peripheral neuropathic pain, NRS numerical rating scale
Table 1 Test and observation scheduleThe study has been approved by the Certified Review Board of Hattori Clinic (CRB3180027; approval date, 20/December/2024) and registered in the Japan Registry of Clinical Trials (jRCTs031240623 registered 20/January/2025). The study will be conducted in accordance with the Ethical Guidelines for Life Science and Medical Research Involving Human Subjects, the Act on the Protection of Personal Information, and the Declaration of Helsinki (as revised in 2013). The study will also be conducted in accordance with the Clinical Trials Act in Japan, under which clinical research is only required to undergo a central ethical review. Written informed consent will be obtained from each patient. No permissions are required for assessment scales other than EQ-5D-5L. The permission for use of EQ-5D-5L was obtained prior to study [29].
Sample SelectionFull detailed eligibility criteria for both study parts are shown in Table 2. Briefly, for the baseline part, patients aged ≥ 18 years with T2DM who provide informed consent will be included.
Table 2 Eligibility criteriaOf the patients enrolled in the baseline survey, those diagnosed with DPNP and with an NRS score for pain of subjective symptoms ≥ 1 will be included in the interventional study. DPN and DPNP will be diagnosed according to the Simple Diagnostic Criteria for Diabetic Polyneuropathy proposed by the Diabetic Neuropathy Study Group in Japan [30, 31]. Subjective symptoms of DPN at the time of secondary enrollment are defined as follows: (1) bilateral; (2) numbness, pain, or abnormal sensation in the toes or soles of the feet (however, abnormal sensation alone is not sufficient). The presence of upper limb symptoms alone is not considered a subjective symptom of DPN in the interventional study. Numbness, pain, and abnormal sensation in the toes and soles of the feet are determined based on the neuropathy questionnaire (see Supplementary methods).
Patients with severe pain due to causes other than DPNP in whom it is considered difficult to assess pain, and patients with creatinine clearance (CrCL) < 30 ml/min will be excluded from the interventional study.
InterventionEligible patients in the interventional study will receive mirogabalin treatment for 12 weeks. Mirogabalin will be administered according to its Japanese package insert and based on each patient’s baseline renal function [26]: for patients with CrCL ≥ 60 ml/min, mirogabalin will be administered at 5 mg twice daily (BID) for the first 2 weeks, 10 mg BID for the next 2 weeks, and 15 mg or 10 mg BID after week 5; and for patients with CrCL 30– < 60 ml/min, the dose will be half that for those with CrCL ≥ 60 ml/min.
The prohibited concomitant medications and therapies in the interventional study are listed in Table 3. The use of concomitant medications will be prohibited during study drug administration, as well as 28 days prior to the second screening. The prohibited therapies will be spinal cord stimulation and any surgical analgesic treatments that might affect efficacy assessment during study drug administration. Changes in the dosage and administration of pain medications taken prior to the start of the interventional study will not be permitted during the study period. Similarly, changes in the frequency of concomitant therapy for pain will not be permitted.
Table 3 Full list of prohibited concomitant medications and therapiesPlanned OutcomesStudy endpoints are shown in Table 4. The primary endpoint is the change in the NRS score for pain from baseline at week 12 in the interventional study. Patients will be asked to rate their most intense pain within the previous 7 days on an 11-point NRS ranging from 0 (“no pain”) to 10 (“worst pain possible”) [32].
Secondary endpoints in the baseline survey include the prevalence of DPN and DPNP, the NRS score for pain in patients diagnosed with DPNP, and the prevalence of each subjective symptom of DPN based on the Simple Diagnostic Criteria for Diabetic Polyneuropathy [30, 31].
Secondary endpoints in the interventional study include: change in the NRS score for pain from baseline at week 4; responder rates at weeks 4 and 12 indexed by ≥ 30% or ≥ 50% reduction in the NRS score for pain; patient impressions of changes in subjective symptoms of DPNP at week 12 (four-point scale: 1 improved, 2 no change, 3 worsened, and 4 no symptoms); patient satisfaction with DPNP treatment at week 12 (seven-point scale: 1 very satisfied, 2 satisfied, 3 slightly satisfied, 4 neither, 5 slightly dissatisfied, 6 dissatisfied, and 7 very unsatisfied); Patient Global Impression of Change (PGIC) score at week 12 (seven-point scale: 1 very much improved, 2 much improved, 3 minimally improved, 4 no change, 5 minimally worse, 6 much worse, and 7 very much worse) [33]; change in the EuroQol-5Dimension-5Level (EQ-5D-5L) score [29] from baseline at weeks 4 and 12; change in the NRS score for sleep disturbance from baseline at weeks 4 and 12 (11-point scale; ranging from 0 “no pain/no sleep disturbance” to 10 “sleep completely disturbed by pain”); and change in the International Physical Activity Questionnaire (IPAQ) from baseline at weeks 4 and 12 [34]. The safety endpoint is adverse events (AEs).
Sample SizeSince study enrollment will be terminated when the number of enrolled patients reaches 100 in the mirogabalin interventional study, the sample size for the baseline survey was set based on that of the interventional study.
First, based on feasibility, the target sample size for the interventional study was set at 100 patients. Assuming a standard deviation (SD) of 1.0–2.0 for the change in the NRS score from baseline to 12 weeks based on the phase 3 trials of mirogabalin [22, 23], and assuming 80–100 subjects in the analysis to consider dropouts from the study, the 95% confidence interval (CI) (one-sided) for the mean change of the NRS score becomes 0.196–0.438.
If the number of cases in the interventional study is 100, the number of cases in the baseline study would be approximately 1000–3000 based on the following assumptions from previous studies [23, 35]: 15–25% of patients with DM have neuropathic pain; 12% of patients are prescribed neuropathic pain medications; 30–50% of patients will newly start DPNP pharmacotherapy in this study. With a baseline caseload of 1000–3000 and DPNP prevalence of 15–25%, the 95% CI (one-sided) for DPNP prevalence becomes 1.3–2.7%.
Data AnalysisFor the baseline survey, the full analysis set (FAS) is defined as all eligible patients. For the interventional study, the FAS is defined as all patients who receive at least one dose of the study drug and have data available from at least one post-dose efficacy evaluation; the per-protocol set (PPS) is defined as all patients in the FAS population who adhere to the study protocol; and the safety analysis set is defined as all patients enrolled in the study who receive at least one dose of the study drug.
For the primary endpoint, the change from baseline at week 12 in the NRS score, the mean, 95% CI, and p-values from paired t tests will be calculated in the FAS. A similar analysis is planned in the PPS as a sensitivity analysis. The same descriptive statistics will also be calculated for the change from baseline at week 4 in the NRS score, the changes from baseline at week 4 and week 12 in the EQ-5D-5L, and the changes from baseline at week 12 in the NRS score for sleep disturbance.
For the prevalence of DPN and DPNP, the proportions of patients and their 95% CIs will be calculated. For the factors associated with the prevalence of DPN and DPNP, univariate and multivariate analyses using logistic regression models are planned to calculate odds ratios and their 95% CIs.
For changes from baseline in IPAQ at week 4 and week 12, summary statistics and interquartile ranges will be calculated, and Wilcoxon's signed-rank test will be used.
AEs will be coded according to the Japanese Medical Dictionary for Regulatory Activities version 27.1 or higher. AEs will be analyzed by severity, and events leading to discontinuation of the study drug will also be analyzed. All statistical analyses will be performed using SAS version 9.4 or higher (SAS Institute Inc.; Cary, NC, USA).
Comments (0)