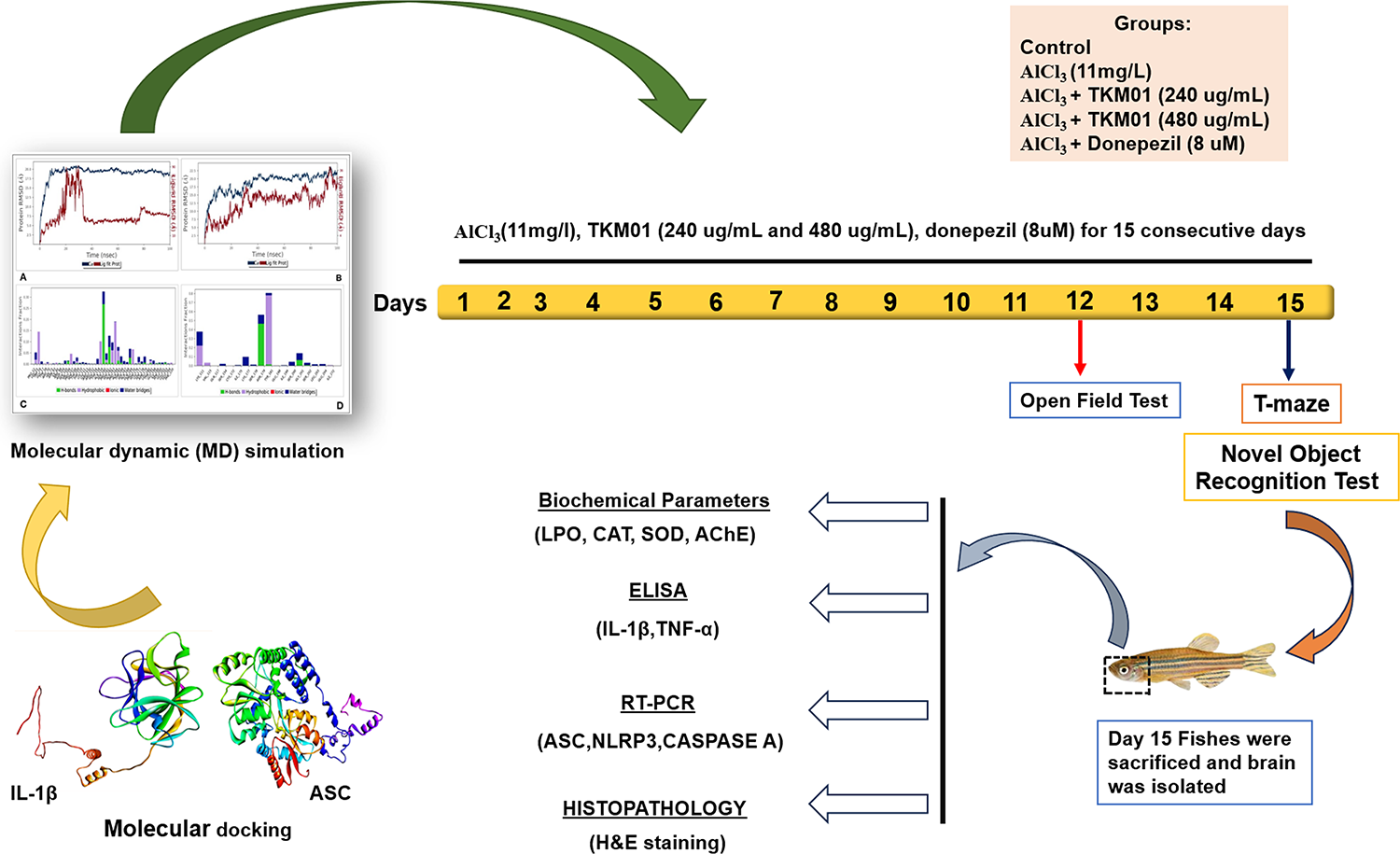
Centratherin
Centratherin was extracted from the leaves of the plant Eremanthus crotonoides (DC.) Sch. Bip., belonging to the Asteraceae family, collected from the Jurubatiba Restinga National Park, Rio de Janeiro, Brazil as previously described [11]. The dried leaves of E. crotonoides were ground and macerated with 98% ethanol to obtain the ethanolic extract. This extract was then partitioned with hexane, generating the hexanic and hydroalcoholic phases. The hydroalcoholic phase was sequentially partitioned with CHCl, from which the CHCl2 extract was obtained. The CHCl2 extract was subsequently analyzed using UPLC-PDA-MS/MS chromatography, allowing its successful identification. To purify centratherin from the CHCl2 extract, fractionation was carried out using flash chromatography under reversed-phase silica gel [12]. The identification of centratherin was confirmed according to previously published 1H and 13C nuclear magnetic resonance (NMR) data in the literature. The 1H NMR spectrum of centratherin (shown in the Fig. S1) confirms its identity and indicates a high level of purity, estimated to be greater than 95%, based on the absence of extraneous resonances. However, a definitive quantification of purity (e.g., by qNMR or HPLC) was not determined at this stage.
Cell Culture
Patient-derived GB cell lines GBM95 and GBM02, as well as cultures of healthy human astrocytes, were established at the Laboratory of Cellular Morphogenesis (Institute of Biomedical Sciences—UFRJ) from tumor specimens obtained at the Clementino Fraga Filho University Hospital, following the ethical guidelines of the Brazilian Ministry of Health, registered with the National Research Ethics Committee under record No. 2340, from 2001. GBM95 and GBM02 cell lines are IDH wild-type, negative for MGMT expression and express the EGFRvIII isoform [13, 14]. Healthy human astrocyte cultures were obtained through a previously established protocol [15], and the derivations of GBM95 and GBM02 from patient tumors were previously described [16]. The U87 cell line was obtained from the American Type Culture Collection (ATCC). All cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with F12 (DMEM-F12—Cat No. 12400—Gibco), supplemented with glucose (35 mM), glutamine (2 mM), penicillin/streptomycin (0.5 mg/mL), fungizone (2.5 μg/mL), and sodium bicarbonate (3 mM) containing 10% Fetal Bovine Serum (FBS—Cat No. 12657—Gibco). The cells were maintained in an incubator at 5% CO2 and 37 °C until they reached confluence. Once confluent, the medium was removed, and 0.5% trypsin/ethylenediaminetetraacetic acid (EDTA) solution (Cat No. 15400—Invitrogen) or 0.02% EDTA (Isofar) in phosphate-buffered saline (PBS—NaCl, Na2HPO4, KH2PO4, and KCl—pH 7.4, 0.1 M) was added to detach the cells, which were then incubated for 5 min. Medium containing serum was added to inactivate trypsin, and the cells were centrifuged for 3 min at 300 g. After centrifugation, the supernatant was discarded, and the pellet was resuspended in medium with serum. The cells were counted in a Neubauer chamber and subsequently plated.
Treatments
Centratherin was diluted in dimethyl sulfoxide (DMSO—Sigma) and then further diluted in medium at final concentrations of 0.5, 1.5, or 2.5 μg/mL (1.3, 4.0, or 6.6 µM). Taxol (Paclitaxel—Cat No. T7402—Sigma), used to induce cell cycle arrest in the M phase, was diluted in DMSO and then in medium at a final concentration of 1 µM. TMZ (Cat No. T2577—Sigma) was diluted in DMSO and then in medium at final concentrations of 100, 500, and 1000 µM. Necrostatin-1 (Nec-1—Cat No. N9037—Sigma) was diluted in DMSO and then in medium at a final concentration of 100 µM for GB cell treatment. The final DMSO concentration, determined by the 2.5 μg/mL centratherin treatment, was 0.001% v/v (~ 140 μM), a biologically insignificant level for cell assays. All control (CTRL) conditions were prepared using this same DMSO concentration, ensuring that any observed effects were attributable to centratherin rather than to the solvent.
MTT Assay
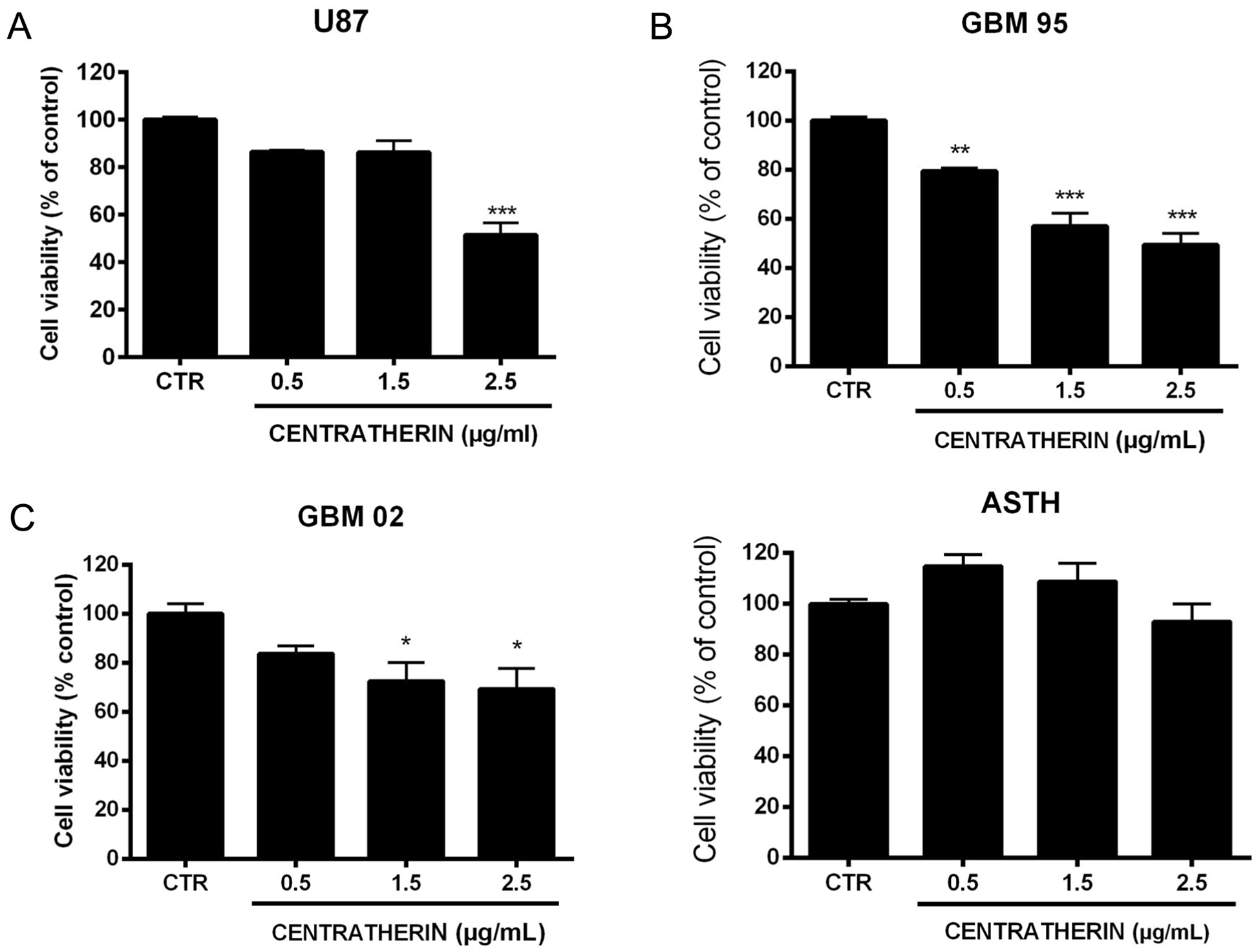
GBM02 and GBM95 cells were plated at a density of 1.5 × 104 cells per well, U87 at 3.0 × 104 cells per well, and healthy human astrocytes at 7 × 103 cells per well in a 96-well plate with medium containing serum and cultured in an incubator at 5% CO2 and 37 °C. Next, cells were treated with centratherin for 24 h. To assess viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT—Sigma—Cat No. M2128) was added to each well at a final concentration of 5 mg/mL and incubated for 2 h at 37 °C. After incubation, the supernatant was removed, 50 µL of DMSO was added to each well, and the plate was shaken for 15 min. Finally, absorbance was quantified using a plate reader (Victor 3 Perkin Elmer) at 570 nm. IC50 values were calculated as previously described [17]. Three independent experiments were performed in triplicate.
Videomicroscopy
GBM02 cells were plated at a density of 4.0 × 105 cells in a 35 mm diameter plate. Then, the cells were transferred to a CO₂ and temperature-controlled incubator (5% and 37 °C, respectively). The following day, the cells were treated with centratherin and placed in another incubator adapted to a Nikon Eclipse TE300 microscope (Nikon) with temperature and CO2 also controlled (37 °C and 5%, respectively). Over the next 20 h, phase-contrast images of the same field for each experimental condition were obtained every minute with the aid of a Hamamatsu C2400 CCD camera (Hamamatsu) attached to the microscope. The resulting image stacks were converted into videos, and analyses of proliferation, motility, and morphological changes were performed using ImageJ software. For proliferation analysis, the number of cells undergoing cytokinesis (the moment when one cell divides into two) in each video frame was quantified. Cell motility was performed as previously described [18]: at least ten cells per condition in each video were randomly selected, their positions tracked from the first to the last frame, and the trajectory traveled (in micrometers) was used to calculate cell speed. Morphological analyses were performed by counting cells with a round morphology in each video frame, as previously described [19]. To analyze the effect of centratherin on proliferation, migration, and morphology of GB cells, at least two independent videos were analyzed for each experimental condition.
3D Invasion Assay
GBM02 cells at a density of 2.0 × 106 cells were plated in serum-containing medium in a non-adherent plate and immediately transferred to a shaker inside a CO₂ and temperature-controlled incubator (5% and 37 °C, respectively) to allow spheroid formation. The following day, these spheroids were plated as previously described [20]. For this, one spheroid was added to each well of a 96-well plate containing type I collagen matrix (serum-free medium, 1 mg/mL collagen I (BD Biosciences—Cat No. 354236), and 7.5 × 10⁻3 M NaOH). Centratherin treatment was added to each well, and at 0, 24, and 48 h, images of each well were acquired using the Nikon Eclipse TE300 microscope (Nikon). Using ImageJ software, the area occupied by the cells at 0, 24, and 48 h was quantified. Cell invasion analysis 24 and 48 h after treatment was performed by normalizing the occupied area to the time 0 measurement. Three independent experiments were conducted in triplicate.
Cell Cycle
GBM02 cells were plated at a density of 4.0 × 105 cells per well in a 6-well plate with serum-containing medium, cultured in an incubator at 5% CO2 and 37 °C, and treated with centratherin the following day. At the end of the 24 h treatment, the supernatant from the wells was collected to harvest the cells that detached during treatment. The remaining cells were washed with PBS and then trypsinized. The trypsinized cells were then added to the tube containing the previously collected supernatant, centrifuged, and washed twice with PBS. Cell fixation was performed with 70% ethanol, as previously described [21]. The tube containing the cell pellet was placed on a vortex, and while being agitated, 1 mL of fixative was added drop wise. The fixed cells were then stored at − 20 °C. For flow cytometry analysis, cells were washed with PBS and incubated with 500 μL of FxCycle™ PI/RNase Staining Solution (Cat No. F10797—Invitrogen) for 30 min at room temperature in the dark. After incubation, cells were analyzed using the Attune NxT flow cytometer (ThermoFisher). The forward scatter (FSC) and side scatter (SSC) parameters were adjusted to locate the cells, and the population of interest was selected by excluding cellular debris. Then, cell doublets were eliminated from the analysis by selecting the population using FSC-area vs. FSC-height parameters. Finally, the PI fluorescence intensity, proportional to the amount of DNA in the cells, was analyzed using a histogram. The histogram displayed two peaks whereas the first peak corresponded to G1 phase cells; the second peak corresponded to G2 or M phase cells, and cells located between the two peaks were identified as being in the S phase of the cell cycle, as described in other studies [22]. Three independent experiments were conducted.
Immunofluorescence
A total of 4.0 × 105 GBM02 cells were plated in a 6-well plate with coverslips and cultured in an incubator at 5% CO₂ and 37 °C. The following day, the cells were treated with centratherin, and at the end of the treatment, cells were fixed with 4% paraformaldehyde (PFA—Isofar) for 15 min or with methanol for 5 min followed by 15 min with 4% PFA (for α-tubulin staining). After fixation, the cells were washed with PBS (pH 7.4, 0.1 M) and permeabilized with 0.2% Triton X-100 (Sigma) for 5 min. Next, nonspecific binding sites were blocked using 5% bovine serum albumin (BSA—Cat No. E588—AMRESCO) for 30 min. After blocking, the cells were incubated in a humid chamber with the primary antibody diluted in 1% BSA overnight at 4 °C or for 1 h at room temperature. After incubation, the cells were washed with PBS and incubated for 2 h in a humid chamber with the secondary antibody and/or Phalloidin, also diluted in 1% BSA. The cells were then washed and incubated with 4′,6′-diamino-2-phenylindole (DAPI) for 5 min to stain the nuclei. After another washing step, the slides were mounted with mounting medium (Fluoromount G). The stainings were visualized using a confocal microscope, and the images were analyzed using ImageJ software. For γH2AX analysis, fluorescence intensity was measured and normalized to the total number of cells in each image. For cytoskeleton analysis—including phalloidin, α-tubulin, and vimentin—filament organization was evaluated by quantifying anisotropy, as previously described [23, 24], using the FibrilTool plugin.
Scanning Electron Microscopy
GBM02 cells were plated at a density of 4.0 × 105 per well in 6-well plates with coverslips and cultured for 24 h in an incubator at 5% CO₂ and 37 °C. The following day, the cells were treated. At the end of the 24 h treatment, the cells were fixed for 1 h with a solution containing 2.5% glutaraldehyde diluted in 0.1 M sodium cacodylate buffer. The cells were then washed with 0.1 M sodium cacodylate buffer and post-fixed with 1% osmium tetroxide diluted in 0.1 M sodium cacodylate for 40 min in the dark. After incubation, the cells were washed again with 0.1 M sodium cacodylate buffer and dehydrated in a graded ethanol series (10–100%). Finally, the samples were transferred to a critical point drying apparatus (Bal-Tec), gold-coated using a metalizer (Bal-Tec), and then analyzed under a Quanta 250 scanning electron microscope (FEI Company), where micrographs were obtained. For scanning electron microscopy analysis, two independent experiments were performed.
Transmission Electron Microscopy
1.7 × 106 GBM02 cells were plated in 100 mm diameter plates. The cells were then treated. After the 24 h treatment, the cells were fixed for 2 h with a solution containing 4% PFA, 2.5% glutaraldehyde, and 0.1 M sodium cacodylate buffer, and washed with PBS pH 7.4, 0.1 M. The cells were then carefully scraped from the bottom of the plate and post-fixed with 1% osmium for 40 min in the dark. The pellet was washed with PBS and dehydrated in a series of increasing acetone concentrations (30–100%) and then infiltrated with a 1:1 acetone: EPON mixture overnight. Subsequently, the pellet was embedded in pure EPON, and the blocks were polymerized at 60 °C for 48 h. 70 nm sections were cut using an ultramicrotome (Leica EM UC7), contrasted with 2% uranyl acetate and 1% lead citrate, and visualized under a Spirit transmission electron microscope. One experiment was conducted for transmission electron microscopy analysis.
LIVE/DEAD Assay
4.0 × 105 GBM02 cells were plated per well in a 6-well plate. The following day, the cells were treated with centratherin, and at the end of the 24 h treatment, the cells were detached with trypsin, washed with serum-free medium, and then incubated with the dye from the LIVE/DEAD™ Fixable Green Dead Cell Stain Kit (Cat No. L23101—Invitrogen) for 30 min on ice in the dark. At the end of the incubation, the cells were washed with 1% BSA diluted in PBS and analyzed on the Attune NxT flow cytometer (ThermoFisher). The parameters of FSC vs. SSC were used to select the population of interest, excluding the cell debris, and the FSC-area vs FSC-height parameters were used to eliminate cell doublets. Histograms of the green dye were analyzed to identify live cells (negative for the dye)—the first peak of the histogram, and dead cells (positive for the dye)—the second peak. Three independent LIVE/DEAD experiments were performed.
Annexin/PI Assay
4.0 × 105 cells were plated in 6-well plate and cultured in an incubator with 5% CO2 at 37 °C. The following day, the cells were treated with centratherin, and at the end of the 24 h treatment, the supernatant from the wells was collected, the cells were trypsinized, and added to a tube with the collected supernatant. The cells were then centrifuged, washed with PBS buffer, resuspended in 96 μL of Annexin V buffer, and incubated with 1 μL of Annexin V and 12.5 μL of Propidium Iodide (PI) from the Annexin V FITC Early Apoptosis Detection Kit (Cat No. #6592—Cell Signaling) for 15 min on ice. After incubation, 300 μL of buffer was added, and the fluorescence was analyzed using the Canto II flow cytometer (BD Biosciences). The FSC vs. SSC parameters were used to select the population of interest, excluding cellular debris, and the FSC-area vs. FSC-height parameters were applied to eliminate cell doublets. Dot plot graphs of FITC and PI fluorescence were created to evaluate Annexin and PI staining for cell death analysis. Cells that are double-negative for Annexin V and PI are considered alive; cells positive only for Annexin V are in the early phase of apoptosis; cells positive only for PI are undergoing necrosis; finally, cells double-positive for Annexin V and PI may be considered in late apoptosis or necrosis [25]. Three independent Annexin/PI assays were conducted to assess the effects of centratherin on GBM cell death.
Protein Extraction
GBM02 cells were plated at 4.0 × 105 cells per well density in 6-well plates and cultured in an incubator with 5% CO2 at 37 °C. The following day, the cells were treated. After treatment, the supernatant from the wells was collected, and the cells were trypsinized. The trypsinized cells were added to a tube containing the collected supernatant, then centrifuged and washed with PBS. After washing, the cell pellet was resuspended in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris HCl pH 8.0, 150 mM NaCl, 1% NP40, 0.5% NaDeoxycholate, 0.1% SDS pH 7.5) in the presence of a protease inhibitor (Sigma—Cat No. P8340) or in tris-urea buffer (UTB) (75 mM TrisHCl, 9 M Urea, 1% Beta-mercaptoethanol). The sample was sonicated (2 pulses of 5 s with a 5 s interval) and then centrifuged at 8000×g for 10 min. After centrifugation, the supernatant was collected, sample buffer (Beta-mercaptoethanol, SDS, Tris 1 M pH 6.8, Glycerol, 0.1% bromophenol blue) was added, and the protein extract was boiled for 5 min at 95 °C.
Western Blot
The protein samples were subjected to polyacrylamide gel electrophoresis (10% or 12%) at 110 V to separate proteins by molecular weight. The proteins were then transferred for 1.5 h at 0.1 A to polyvinylidene fluoride (PVDF) membrane (Bio-Rad), activated with methanol for 5 min. The membranes were incubated with 5% skim milk (Molico) for 1 h at room temperature under agitation to block non-specific binding sites and then incubated overnight at 4 °C with the primary antibody diluted in 1% milk. After incubation, the membranes were washed with tris-buffered saline (TBS) containing Tween-20 (TBST) (NaCl 150 mM, Tris 50 mM, Tween 20 0.1%, pH 7.5) and then incubated for 1 h and 20 min with the secondary antibody diluted in 1% milk. The membranes were washed with TBST. The bands were visualized by fluorescence emission using the Odyssey apparatus (LICor). The densitometry of the bands was performed using the UN-SCAN IT software. Three independent experiments were performed for the western blot analyses.
Statistical Analysis
All values were expressed as mean ± standard error, except for the cell motility analysis performed through videomicroscopy (which was expressed in the form of a box plot). Statistical analysis of the results was performed using one-way ANOVA followed by Dunnett’s post-test using the GraphPad Prism software, comparing the experimental groups with the control. p Values < 0.05 were considered significant (*p < 0.05; **p < 0.01; ***p < 0.001).

Comments (0)