
Remember me
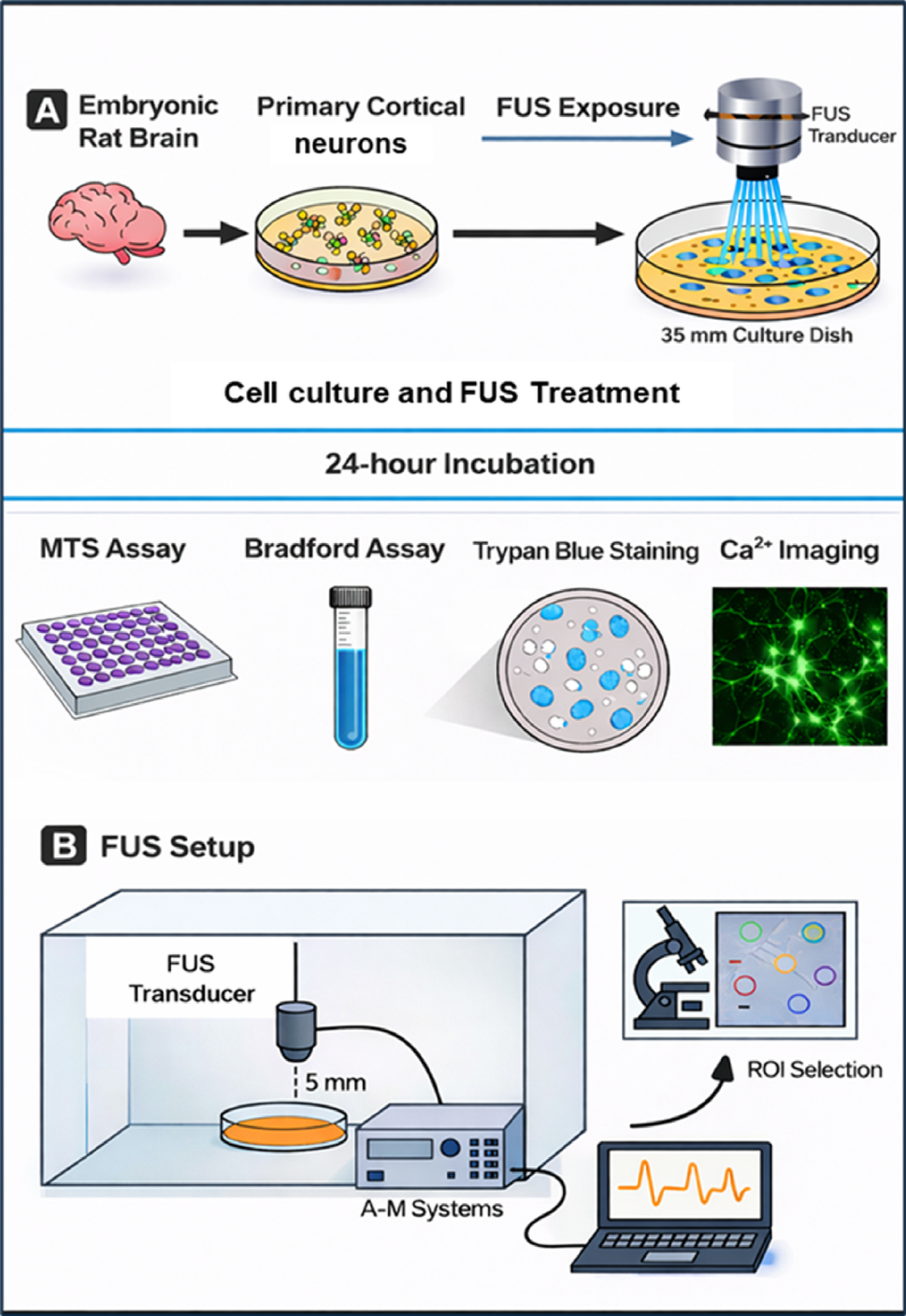
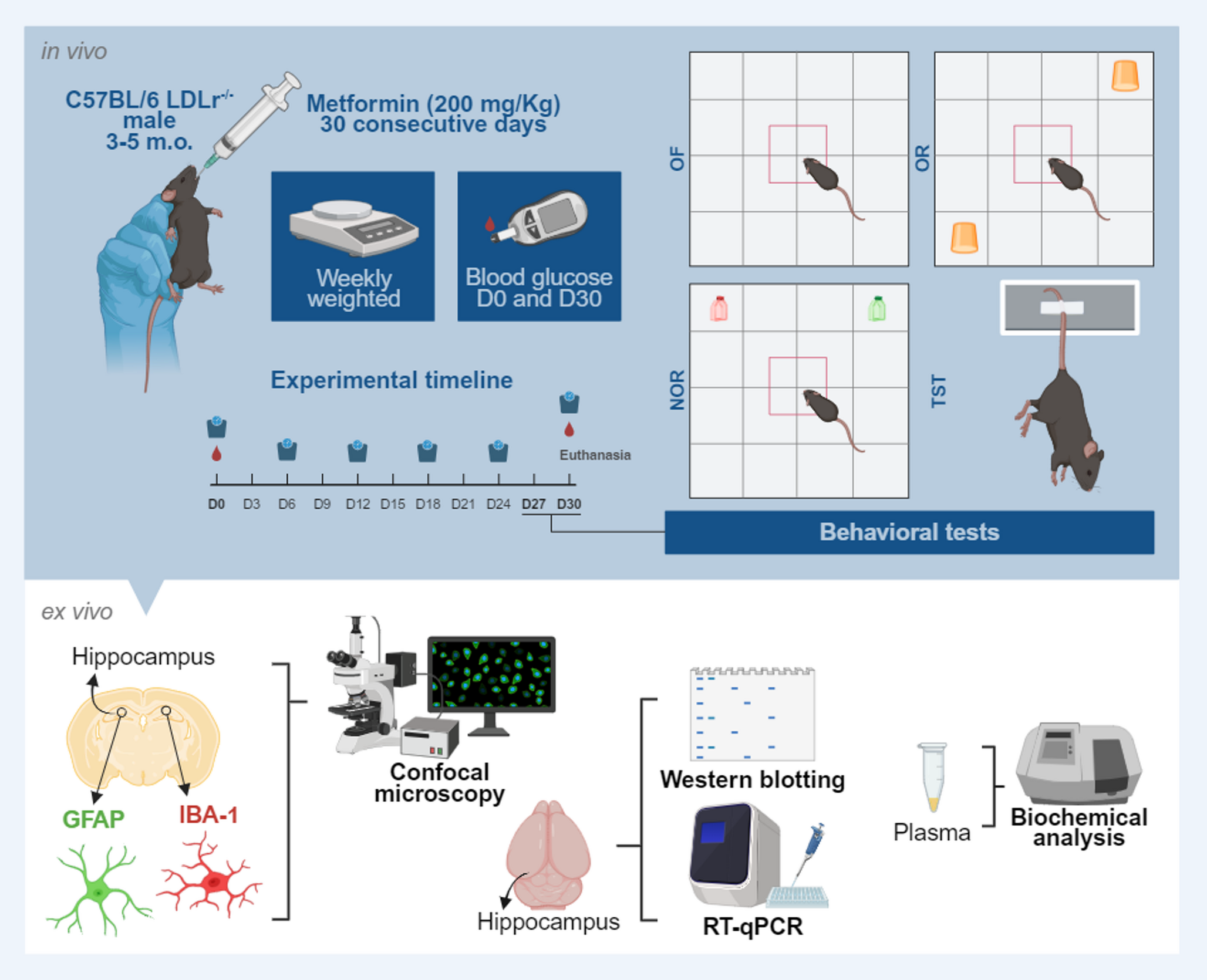
The experimental procedures involving animals were approved by the Ethics Committee of the National Institute of Mental Health. The dissection and isolation of cortical cells from rat embryos (day 18) was performed by following the Jorratt et al. (2022) [15]. Briefly, pregnant Wistar rats (approximately 3–4 months old, 250–300 g body weight) were sacrificed via cervical dislocation, in accordance with established ethical guidelines for animal research. Embryonic brains were carefully extracted using sterilized, autoclaved surgical instruments under aseptic conditions to prevent contamination. The cortical regions were isolated in a 15 ml conical tube and washed thrice with ice-cold Hank’s Balanced Salt Solution (HBSS; Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. 14175095). Then, they were transferred to a 1.5 mL microcentrifuge tube and triturated using a 0.9 mm needle (B. Braun, 4657519) and a 0.45 mm needle (B. Braun, 4657683) three times each, at a pace of one drop per second. The tissue suspension was filtered through a 100-µm pore nylon filter to remove undissociated tissue fragments and debris. The filtered suspension was collected into a seeding medium composed of Dulbecco’s Modified Eagle Medium (DMEM; Biowest, Nuaillé, France; Cat. No. L0104-500), supplemented with 10% fetal bovine serum (FBS; Biowest, Nuaillé, France; Cat. No. S1810) and 1% penicillin-streptomycin (1%; Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. 15070063), to provide essential nutrients and maintain sterility for optimal cell viability and growth. Cells were counted using a hemocytometer, and dilutions were prepared according to the experiment: 125,000 cells/cm² were plated for the MTS assay and around 50,000 cells/cm2 for Ca2+ imaging in 35 mm petri dishes (Cellvis, Mountain View, CA, USA; Cat. No. D35-14-1.5.5-N) coated with Poly-L-lysine. Cultures were incubated at 37 °C and 5% CO2 for 24 h. The next day, the seeding medium was replaced with a growth medium, which contained Neuronal medium with 1% penicillin-streptomycin (1%; Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. 15070063), 2 mM L-Glutamine (Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. 25030149), and 2% B27™ serum-free supplement (Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. A3582801). Every 4–5 days, the growth medium was replaced with a fresh medium.
Application of FUSFUS was applied on day 14 in vitro, and the grown cells were divided into two experimental groups named Control and FUS. The Control group did not receive any ultrasound stimulation. In contrast, the FUS group was subjected to FUS at +5 V and +10 V for 10 min using a 300 kHz custom immersion-type transducer compatible with the A-M Systems 4100 High-Power Stimulator. Stimulation strength is reported in terms of applied electrical drive voltage (+5 V and +10 V), which serves as a relative measure specific to this experimental setup, as direct acoustic pressure or intensity calibration of the transducer was not performed. The transducer was sterilized before use through a combination of 70% ethanol treatment and 40 min of UV light exposure. During the experiments, the transducer was positioned vertically (at a 90° orientation) at a fixed distance of 5 mm from the base of the Petri dish. At this distance, the ultrasound field uniformly covered the entire 35-mm culture area; therefore, all imaged cells were located within the insonated region. Hence, under these conditions, the ultrasound beam encompassed the entire culture area, and all subsequent cell viability assays and Ca2+ imaging analyses were performed on cells located within the insonated region. During confocal imaging, specific ROIs were manually selected in each FUS-exposed dish to quantify intracellular Ca2+ responses. The stimulation parameters included a 300 kHz monophasic pulse with a duration of 0.5 s, a train burst count of 30, and a period of 20 s (Table 1). During stimulation, petri dishes were maintained at 37 °C using a plate heater, and the experimental setup was housed within a laminar flow hood to ensure sterility. After each stimulation session, the transducer was removed, sanitized with 70% ethanol, and reused for subsequent cultures. Following FUS treatment, cells were returned to the incubator for 24 h. In the Control group, cells underwent the same process of transducer submersion and withdrawal without exposure to FUS, ensuring consistency across experimental conditions (Fig. 1).
Fig. 1
Experimental schematic of FUS in primary cortical neurons. A Primary cortical neurons (DIV14) cultured in 35 mm dishes were exposed to low-intensity pulsed FUS (300 kHz, 10 min; 5–10 V drive voltage) using a transducer positioned 5 mm above the culture dish. Cell viability, protein content, morphology, and intracellular Ca2+ signaling were assessed 24 h after FUS exposure. B FUS stimulation apparatus and Ca2+ imaging workflow. A 300 kHz transducer was positioned 5 mm above the culture dish containing primary cortical neurons in pink medium, maintained at 37 °C inside a sterile laminar hood. The transducer was driven by an A-M Systems Model 4100 isolated high-power stimulator connected to a monophasic waveform generator and data-acquisition laptop. The insect illustrates regions of interest (ROIs) selection during confocal Ca2+ imaging, where individual somata were analyzed for fluorescence intensity changes following FUS exposure. All ROIs used for functional analysis were selected from areas directly exposed to the ultrasound field
Table 1 FUS experimental protocol and parameter settings programmed on the A-M systems (Sequim, WA, USA) model 4100 isolated High-Power stimulatorTotal Protein QuantificationFollowing FUS stimulation and incubation for 24 h, cell lysates from each treatment group (Control, FUS 5 V, and FUS 10 V) were collected for total protein quantification. The culture medium was removed, and cells were rinsed twice with cold phosphate-buffered saline (PBS). Neuronal lysates were prepared by adding ice-cold radioimmunoprecipitation assay (RIPA buffer; MilliporeSigma, Burlington, MA, USA; Cat. No. 20–188) and incubating on ice for 20 min with intermittent vortexing. The lysates were centrifuged at 12,000 × g for 10 min at 4 °C, and the supernatants were used for protein measurement. Protein concentration was determined using the Bradford protein assay (Bio-Rad Protein Assay Kit, USA) with bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA; Cat. No. A9647) as the standard. A series of BSA standards (0–1.5 mg/mL) was prepared to generate a calibration curve. For each sample, 10 µL of diluted lysate was mixed with 200 µL of Bradford reagent in a 96-well plate, incubated for 5 min at room temperature, and the absorbance was recorded at 595 nm using a microplate reader (Tecan Infinite M200 Pro). All samples were analyzed in triplicate, and protein concentrations were calculated from the BSA standard curve, accounting for the dilution factor, and expressed as µg/mL.
MTS Cell Viability AssayCell viability was evaluated using the 3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)−2 H-tetrazolium (MTS; Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. L11939.03), also known as the MTS colorimetric assay, which is based on the reduction of the MTS tetrazolium compound to generate a formazan product within living cells. Cultures were treated with FUS for 10 min and later incubated for 24 h at 37 °C. After 24 h, 10 µL of MTS solution was added to 100 µL of growth medium and incubated for 2 h at 37 °C with 5% CO2. Triton was used as a positive control. After shaking for 20 s, the optical density (OD) was measured at 490 nm using a plate reader (Tecan Infinite M200 Pro). Data was obtained from 5 biological replicas and expressed as a percentage of cell viability using the formula: (OD sample/OD control) × 100. Additionally, trypan blue was used to visualize cell viability in cultures. The growth medium was replaced with 0.02% trypan blue (Gibco, Thermo Fisher Scientific, USA: 15250061) in PBS for 10 min and then washed three times with PBS for 5 min each. Pictures were taken under the inverted phase contrast microscopy.
Ca2+ Imaging and Confocal MicroscopyFor Ca2+ imaging, primary cortical neurons were divided into Control and FUS-treated groups. Ca2+ imaging was performed using the 10 V condition only (selected as the higher stimulation setting); therefore, voltage-dependent differences in Ca2+ responses between 5 V and 10 V are not reported. Cells in the FUS-treated group were exposed to low-intensity pulsed ultrasound at 10 V for 10 min, while control cells did not receive FUS but were kept under identical conditions inside the biosafety cabinet for the same duration of time. All Ca2+ imaging experiments were performed 24 h after FUS exposure (offline assessment); no real-time Ca2+ recordings were acquired during FUS delivery. After FUS treatment, both groups were incubated with the Ca2+ -sensitive fluorescent dye Fluo-3 AM (Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. F1241) for 1 h at 37 °C in the dark. Following incubation, cells were washed three times with PBS and allowed to stand for 30 min to enable complete de-esterification of the dye. Fresh media was added, and Ca2+ imaging was performed using a Leica TCS SP8X confocal microscope. During live-cell recording, 200 µL of 1 mM KCl was added at the 60-second mark to induce Ca2+ influx through voltage-gated Ca2+ channels, as a positive control for neuronal responsiveness. Time-lapse recordings were acquired continuously for 8 min to capture dynamic changes in intracellular Ca2+ levels, expressed as relative fluorescence intensity (F/F₀).
Comments (0)