
Remember me
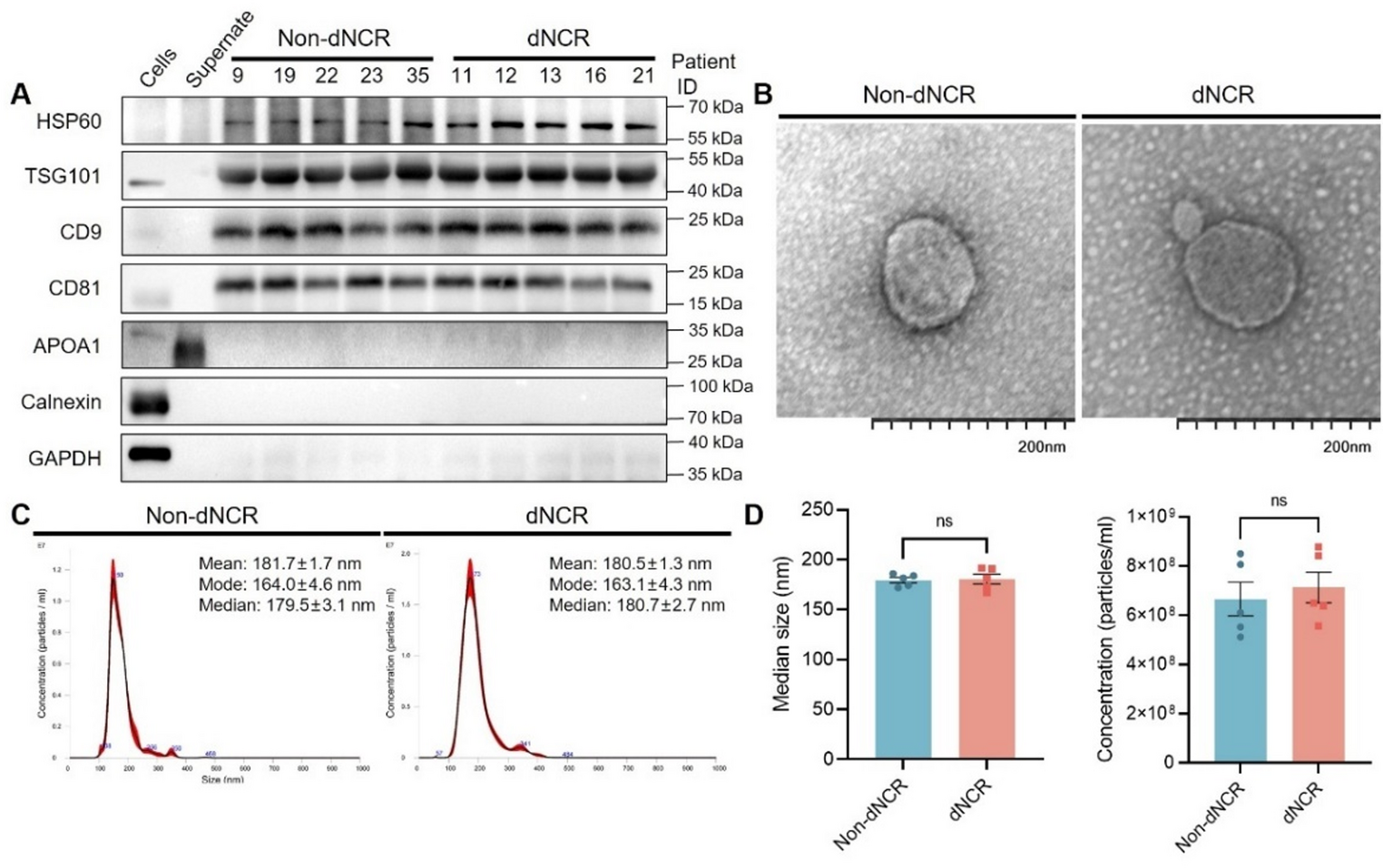
The plasma samples used in this study were selected from our previously established cohort (Zhang et al., PMID: 36457760). From this cohort, we selected two matched sets through strict age-matching criteria (± 3 years): a discovery cohort consisting of 5 patients with dNCR and 5 non-dNCR controls, and a validation cohort comprising 15 dNCR and 15 non-dNCR patients (clinical characteristics are summarized in Tables 1 and 2, respectively). All participants met the original inclusion and exclusion criteria of the parent study.
Inclusion criteria were as follows: (1) age 65 years or older; (2) preoperative Mini-Mental State Examination (MMSE) score of 20 or higher; (3) scheduled for elective non-cardiac surgery under general anesthesia; and (4) American Society of Anesthesiologists (ASA) physical status I–III. Exclusion criteria included: (1) history of cerebrovascular disease; (2) current use of sedative or antidepressant medications; (3) diagnosis of severe neurological or psychiatric disorders; (4) significant auditory or visual impairment that could compromise cognitive assessments; (5) unwillingness to comply with the study protocol or subsequent withdrawal; (6) postoperative admission to the intensive care unit (ICU); and (7) occurrence of postoperative delirium (POD). A diagnosis of dNCR was defined as a decline of ≥ 1 standard deviation (equivalent to a Z-score ≥ 1) from the preoperative baseline in two or more cognitive domains. Detailed patient characteristics and scale scores for all participants are summarized in Supplementary Material 1.
All samples were collected on the 7th day post-operation after the diagnosis of dNCR using standardized protocols: venous blood was drawn into ethylenediaminetetraacetic acid (EDTA) tubes, processed within 2 hours through double differential centrifugation (3,000 g × 15 minutes followed by 12,000 g × 30 minutes at 4 °C), and stored at −80 °C until analysis. Samples were aliquoted to prevent freeze-thaw cycles.
Research involving human subjects has been evaluated and approved by the Institutional Review Board of Tongji University (IRB2021-LCYJ-014) and registered with the Chinese Clinical Trial Registry (ChiCTR) under the unique identifier ChiCTR2100043475 (www.chictr.org.cn). Participants provided written consent to participate in the study.
Hemolysis ControlHemolysis was evaluated in plasma samples using two established methods to ensure sample quality. Free hemoglobin concentration was measured by spectrophotometry using a NanoDrop 2000/2000c instrument, following an established method for hemolysis assessment in plasma [27,28,29,30]. A calibration series was generated by spiking control plasma with lysates from washed human red blood cells (lysed in ddH2O for 30 minutes at room temperature), producing final hemolysis levels of 0.0%, 0.008%, 0.016%, 0.031%, 0.0625%, and 0.125% (v/v). Absorbance was measured at 414 nm (Soret band for heme), 450 nm (bilirubin correction), and 700 nm (turbidity correction). Hemoglobin concentration (mg/dL) was calculated for each study sample using the formula: Hb = 154.7 × A414 − 130.7 × A450 − 123.9 × A700. All plasma samples used in subsequent analyses contained ≤ 20 mg/dL hemoglobin. In parallel, a miRNA-based hemolysis assessment was performed according to the method described by Chico-Sordo et al. [31]. Expression levels of hsa-miR-451a and hsa-miR-23a-3p were measured in duplicate by RT‑qPCR, and a ΔCq value was calculated as mean Cq (hsa-miR-23a-3p) - mean Cq (hsa-miR-451a) for each sample. Higher ΔCq values correspond to a higher degree of hemolysis. Detailed hemolysis data, including calculated hemoglobin concentrations and ΔCq values for all samples, are provided in Supplementary Material 2.
Isolation and Characterization of Plasma ExosomesIsolation of Plasma ExosomesPlasma-derived exosomes were isolated using differential centrifugation. Briefly, frozen plasma samples were thawed at 37 °C and centrifuged at 2,000 g (4 °C, 30 minutes) to remove cellular debris. The supernatant was further clarified by centrifugation at 10,000 g (4 °C, 45 minutes) to eliminate large vesicles, followed by filtration through a 0.45 μm membrane. The filtrate was subjected to ultracentrifugation at 100,000 g (4 °C, 70 minutes) to pellet exosomes. The pellet was washed with 10 mL cold phosphate buffer saline (PBS, PB180327, Procell, China) and centrifuged again under identical conditions. Purified exosomes were resuspended in 100 µL PBS and stored at −80 °C for downstream analyses.
Nanoparticle Tracking Analysis (NTA)Exosome size and concentration were quantified using NTA (NanoSight NS300, UK) featuring an sCMOS camera and Blue488 laser excitation. Each exosome sample was diluted 5000-fold in double distilled H2O (ddH2O) immediately prior to measurement. For every diluted sample, we captured five independent 60-second videos to ensure statistical reliability. The camera settings were maintained at consistent levels: camera level 10–11, shutter speed 696–890, gain 73–146, and frame rate of 25.0 frames per second (1498 frames per video). All measurements were performed under controlled conditions: temperature 22–24 °C, viscosity 0.922–0.936 cP (water reference), and constant syringe pump speed of 100. Particle size parameters—including mean, mode, and median diameter—along with particle concentration, were calculated using the NTA software (version 3.2) (summarized in Supplementary Material 3).
Transmission Electron Microscope (TEM)Exosome morphology was analyzed using the following optimized TEM protocol: Freshly thawed exosome samples were placed on ice and gently vortexed after complete thawing. The supernatant was adjusted to an appropriate concentration with PBS ( PB180327, Procell, China) (pH 7.4). Using precision tweezers, 15 µL of exosome suspension was carefully loaded onto 200-mesh copper grids (NC0859496, Electron Microscopy Sciences, USA) and allowed to adsorb for 1 minute at room temperature. Excess liquid was carefully absorbed with filter paper without touching the grid surface. Negative staining was performed by applying 15 µL of 2% uranyl acetate (201030, Sigma-Aldrich, USA) for 1 minute at room temperature. For samples with visible particulate contamination, the grids were gently washed with ultrapure water using a drop-and-blot technique repeated 2–3 times. After complete drying at room temperature for 15 minutes in a dust-free environment, samples were examined using a JEM-2100 TEM (JEOL, Japan) operated at 80 kV. Images were captured at 80,000× magnification using a Gatan Orius SC1000 CCD camera under standard operating conditions.
Exosomal Western BlottingExosomes were homogenized in M-PER Protein Extraction Buffer (78501, Thermo Fisher Scientific, USA) containing a protease inhibitor cocktail (P8340, Sigma-Aldrich, USA) on ice for 30 minutes. Supernatants’ protein concentrations were determined using BCA Protein Assay Kit (23227, Pierce, USA). For exosome samples, 10 µg of protein was loaded per lane. Protein samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (P0015, Beyotime, China) at 90 V for 2 hours with a 12.5% gel (P0015, Beyotime, China) using a Mini-PROTEAN Tetra System (Bio-Rad, USA). Resolved proteins were then electrotransferred onto polyvinylidene difluoride (PVDF) membranes (0.22 μm, STM2005, Invitrogen, USA) at 90 V for 100 minutes in a transfer buffer containing 25 mM Tris, 192 mM glycine, and 20% methanol. Membranes were blocked with 5% non-fat milk in Tris-buffered saline with 0.1% Tween-20 (TBST, PS103S, Epizyme, China) for 1.5 hours at room temperature before immunoblotting. Immunoblots were probed with purified primary antibodies for HSP60 (ab190828, rabbit, 1:1000, Abcam, USA), TSG101 (ab133586, rabbit, 1:2000, Abcam, USA), CD81 (ab155760, rabbit, 1:1000, Abcam, USA), CD9 (PC22293, rabbit, 1:1000, Abmart, China), ALIX (T57215, rabbit, 1:1000, Abmart, China), APOA1 (DF6264, rabbit, 1:500, Affinity Biosciences, China), Calnexin (ab92573, rabbit, 1:1000, Abcam, USA) or GAPDH (33033, mouse, 1:2000, Bioss, China) overnight at 4 °C, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse (A4416, 1:10,000) or goat anti-rabbit (A0545, 1:10,000) secondary antibodies (Cell Signaling Technologies, USA). Immunoblotting was done with Pierce ECL Western Blotting Substrate. Signals were obtained by Tanon 5200 Multi intelligent imaging system (Tanon, Shanghai, China). Uncropped Western blot images have been provided in the Supplementary Material 4 to ensure complete transparency.
Exosomal miRNA Sequencing AnalysisPlasma-derived exosomal microRNA was extracted using the exoRNeasy Serum/Plasma Kit (77023, Qiagen, Germany) in accordance with the manufacturer’s protocol. Briefly, exosome lysates were homogenized with QIAzol Lysis Reagent (79306, Qiagen, Germany) and chloroform (PHR1552, Merck, Germany), after which RNA precipitation was performed using ethanol. The RNA was purified through exoEasay affinity membrane centrifugal column with Buffer RWT and RPE washes, and subsequently eluted in RNase-free water. RNA quality was assessed utilizing a Quantus Fluorometer and Qsep100 analysis. Small RNA libraries were prepared employing a PE150 sequencing strategy, followed by quality control using FastQC. Raw reads were processed with Fastp for adapter trimming and quality filtering, applying a Q20 threshold. The processed sequences were aligned against the Rfam database using Bowtie to eliminate non-coding RNAs (rRNA/tRNA), followed by miRNA quantification with miRDeep2 and differential expression analysis using DESeq2. For subsequent analysis, miRNAs were prioritized if they met three or more of the following criteria: (1) adjusted p-value (q-value) < 0.05; (2) mean expression (TPM) > 2 in at least one group (dNCR or non-dNCR); (3) absolute fold change (FC) > 2 between groups; (4) detectable expression (TPM > 0) in at least one sample per group; and (5) annotation as known miRNAs in miRBase to facilitate mechanistic validation. Predicted target genes of these prioritized miRNAs were subsequently subjected to functional enrichment analysis based on the Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome pathway databases, with a significance threshold of p < 0.05. Data generated by exosomal microRNA sequencing have been deposited in the SRA repository under accession numbers: SRA: PRJNA1304166.
Cell Culture and TreatmentThe SH-SY5Y cell line was obtained from the American Type Culture Collection (ATCC, USA). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, 11965118, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, 26140079, Gibco, USA), 100 U/mL penicillin, and 100 U/mL streptomycin (PB180120, Pricella, China), and maintained at 37 °C in a 5% CO2 humidified atmosphere. Cell viability was determined using a Cell Counting Kit-8 (CCK-8, C0039, Beyotime, China). All cells used in experiments were within 20 passages. For drug treatment experiments, cells were seeded in 12-well plates (1 × 105 cells per well) and 24 hours after transfection, treated with Ferrostatin-1 (Fer-1, HY-100579, MCE, China) or Erastin (HY-15763, MCE, China) at 5 µM for 24 hours.
Cell TransfectionSH-SY5Y cells were seeded in 12-well plates at densities of 1 × 105 cells per well, and cultured until 50–70% confluence. For hsa-miR-3677-3p modulation, cells were transfected using HiPerFect Transfection Reagent (301705, Qiagen, Germany) with 50 nM hsa-miR-3677-3p mimics or mimic negative control (NC), or 100 nM hsa-miR-3677-3p inhibitor or inhibitor negative control (inhibitor-NC). For ABCB8 knockdown, transfection was performed using Lipofectamine 3000 (L3000015, Thermo Fisher Scientific, USA) with 50 nM siRNA targeting ABCB8 (si-ABCB8), negative control (si-NC), or siRNA targeting GAPDH (si-GAPDH) as a positive control. All oligonucleotides were synthesized by GenePharma (China). After 6 hours of transfection, the medium was replaced with complete culture medium. Cells were harvested at 24 hours post-transfection for RNA analysis, and at 48 hours for protein extraction and functional assays.
Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)Purification of miRNA and total RNA from cells was performed using the miRNeasy Micro Kit (217084, Qiagen, Germany), while exosomal microRNA was isolated from plasma-derived exosomes utilizing the exoRNeasy Serum/Plasma Starter Kit (77023, Qiagen, Germany), following the manufacturer’s protocol. miRNA and total RNA was quantified at NanoDrop 2000/2000c Spectrophotometer (Thermo Fisher Scientific, USA). cDNA was then synthesized through the reverse transcription of mRNA using the Evo M-MLV RT Mix Kit (AG11728, Accurate Biology, China), while the stem-loop method was used for reverse transcription and quantitative analysis of the miRNA (MR201, Vazyme, China). qRT-PCR was performed using miRNA Unimodal SYBR qPCR Master Mix (MQ102, Vazyme, China) and SYBR Green Master Mix (Q712-02, Vazyme, China) on a QuantStudio 6 Flex system, using the following cycling conditions: 95 °C for 30 seconds, followed by 40 cycles of 95 °C for 10 seconds and 60 °C for 30 seconds, with melting curve analysis to confirm amplification specificity. Relative expression levels were calculated using the 2 − ΔΔCt method, with RNU44 or RNU6 (U6) as endogenous controls for miRNA and ACTIN for mRNA. For miRNA quantification, Ct values > 35 were considered undetectable and excluded from analysis; missing data resulting from amplification failures were not imputed. Transcripts were amplified using gene-specific primers (summarized in Supplementary Material 5), which includes both stem-loop RT primers (containing a 5’ universal sequence and 3’ miRNA-specific anchors) and qPCR primers (miRNA-specific forward primers paired with a universal reverse primer) for miRNA and mRNA detection. The raw Ct values from all qRT-PCR analyses are compiled in Supplementary Material 6.
Western BlottingCultured cells were homogenized in M-PER Protein Extraction Buffer (78501, Thermo Fisher Scientific, USA) containing a protease inhibitor cocktail (P8340, Sigma-Aldrich, USA) on ice for 30 minutes, and subsequently centrifuged at 12,000 rpm for 15 minutes at 4 °C. Supernatants’ protein concentrations were determined using BCA Protein Assay Kit (23227, Pierce, USA). For cell lysate samples, 15 µg of protein was loaded per lane. Protein samples were resolved by SDS-PAGE (P0015, Beyotime, China) at 90 V for 2 hours using a 12.5% gel (P0015, Beyotime, China) using a Mini-PROTEAN Tetra System (Bio-Rad, USA). Resolved proteins were then electrotransferred onto PVDF membranes (0.2 μm, STM2005, Invitrogen, USA) at 90 V for 100 minutes in a transfer buffer containing 25 mM Tris, 192 mM glycine, and 20% methanol. Membranes were blocked with 5% non-fat milk in TBST (PS103S, Epizyme, China) for 1.5 hours at room temperature before immunoblotting. Immunoblots were probed with purified primary antibodies for ABCB8 (A2653, rabbit, 1:1000, ABclonal, China), xCT/SLC7A11 (A2413, rabbit, 1:1000, ABclonal, China), GPX4 (A11243, rabbit, 1:1000, ABclonal, China), Total OXPHOS (ab110413, mouse, 1:1,000, Abcam, USA), GAPDH (33033, mouse, 1:2000, Bioss, China) or β-ACTIN (8H10D10, mouse, 1:5,000, Cell Signaling Technologies, USA) overnight at 4 °C, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse (A4416, 1:10,000) or goat anti-rabbit (A0545, 1:10,000) secondary antibodies (Cell Signaling Technologies, USA). Immunoblotting was done with Pierce ECL Western Blotting Substrate. Signals were obtained by Tanon 5200 Multi intelligent imaging system (Tanon, Shanghai, China).
Dual-Luciferase Reporter AssayThe interaction between hsa-miR-3677-3p and ABCB8 was validated using a dual-luciferase reporter assay. DNA fragments spanning the predicted binding site (positions 1586–1592) within the ABCB8 3’UTR were synthesized and cloned into the pmirGLO vector (E1330, Promega, USA) downstream of the firefly luciferase open reading frame, using NheI and SalI restriction sites. The MUT construct incorporated nucleotide substitutions designed to disrupt hsa-miR-3677-3p binding efficacy, thereby serving as a specificity control. 293 T cells were seeded in 6-well plates at a density of 5 × 105 cells per well and co-transfected using Lipofectamine 3000 (L3000015, Thermo Fisher Scientific, USA). Each transfection included either WT or MUT reporter plasmid (3 µg) together with 80 pmol of hsa-miR-3677-3p mimics or negative control miRNA, achieving a final concentration of 80 nM per well. The pmirGLO vector enables simultaneous expression of both Firefly luciferase (Fluc, experimental reporter) and Renilla luciferase (Rluc, internal control), allowing normalization for variations in transfection efficiency and cell viability. At 24- or 48-hours post-transfection, cells were lysed, and luminescence was quantified using the Dual-Luciferase Reporter Assay System (DL101, Vazyme, China). Fluc activity was measured first through oxidation of D-luciferin (emission peak 560 nm), followed by quenching of Fluc signal and subsequent measurement of Rluc activity via coelenterazine oxidation (emission peak 465 nm). Fluc luminescence values were normalized to Rluc activity to account for variations in transfection efficiency. Each experiment was performed in three independent biological replicates.
TEM of CellsCells were fixed with 2.5% glutaraldehyde (PH9003, Phygene, China) in 0.1 M PBS (pH 7.4) for 2 hours at 4 °C, followed by post-fixation with 1% osmium tetroxide (251755, Merck, Germany) for 1 hour. After dehydration through a graded ethanol series (50%, 70%, 90%, and 100%), samples were embedded in epoxy resin (45359, Merck, Germany). Ultrathin Sects. (70–90 nm) were prepared using a Leica EM UC7 ultramicrotome and collected on 200 mesh copper grids (930369, Sigma-Aldrich, USA). Grids were stained with 2% uranyl acetate (15 minutes) and Reynolds’ lead citrate (5 minutes), rinsed with ultrapure water, and air-dried. Samples were imaged using a JEM-2100 TEM (JEOL) at 80 kV, with magnifications ranging from 30,000× (Gatan Orius SC1000 CCD camera).
Mitochondrial Iron DetectionMitochondrial iron levels were assessed using the Mito-FerroGreen probe (Dojindo, M489, Japan). For quantitative measurement, cells were seeded in 96-well clear-bottom plates (Thermo Scientific™ Nunc, 165305) at a density of 1 × 104 cells per well. After treatment, cells were washed twice with HBSS (Gibco, 14175-079) and incubated with 5 µM Mito-FerroGreen in HBSS for 30 minutes at 37 °C in the dark. Following two additional washes, fluorescence was measured using a multimode microplate reader (Infinite 200 Pro, Tecan, Switzerland) with an excitation wavelength of 505 nm and an emission wavelength of 535 nm. Fluorescence intensity was normalized to total protein content or cell count per well.
For imaging, cells were cultured in confocal dishes under identical treatment conditions. After staining with 5 µM Mito-FerroGreen as described above, nuclei were labeled with Hoechst 33,342 (5 µg/mL; Beyotime, C1022) for 10 minutes at 37 °C. Images were acquired using a Keyence BZ-X800 microscope with a 60× oil immersion objective (Keyence, Japan). Mito-FerroGreen was excited at 488 nm and emission collected at 505–535 nm; Hoechst 33,342 was excited at 405 nm with emission at 346–460 nm. Fluorescence intensity was quantified using ImageJ (ImageJ software v1.6.0, NIH, Bethesda, USA).
Malondialdehyde (MDA) ContentCellular MDA levels were assessed using Lipid Peroxidation MDA Assay Kit (S0131S, Beyotime, China). Briefly, SH-SY5Y cells (approximately 1 × 106 cells per sample) were homogenized in PBS and centrifuged (10,000 ×g, 10 minutes, 4 °C) to remove insoluble debris. A total of 50 µL of the supernatant was then transferred and mixed with 100 µL of the kit’s working solution containing thiobarbituric acid (TBA). The mixture was heated in a boiling water bath for 15 minutes to form MDA-TBA adducts. After cooling to room temperature, the absorbance of the pink-colored reaction product was measured spectrophotometrically (VersaMax™ Tunable Microplate Reader, Molecular Devices, USA) at 532 nm.
Ratio of GSH/GSSGThe ratio of Glutathione/Glutathione Disulfide (GSH/GSSG) was determined using a commercial GSH and GSSG Assay Kit (S0053, Beyotime, China). Briefly, cell samples (approximately 1 × 106 cells per sample) were homogenized in ice-cold protein removal reagent M solution (provided in the kit) and centrifuged at 10,000 ×g for 10 minutes at 4 °C. The resulting supernatant was divided into two aliquots: one for total glutathione detection treated with 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB), and the other for GSSG quantification pretreated with 1-methyl-2-vinylpyridinium triflate to scavenge GSH. After incubation at room temperature for 25 minutes, absorbance was measured at 412 nm using a spectrophotometer (VersaMax™ Tunable Microplate Reader, Molecular Devices, USA). The GSH content was calculated as (total glutathione – 2 × GSSG), and the GSH/GSSG ratio was derived to evaluate cellular oxidative stress.
Measurement of Mitochondrial Lipid PeroxidationMitochondrial lipid peroxidation was assessed using the MitoPerOx assay kit (HY-125623, MCE, China). Cells were seeded in Thermo Scientific™ Nunc MicroWell 96-well plates (165305, Thermo Fisher Scientific, USA) with clear bottoms at a density of 1 × 104 cells per well and treated accordingly. After treatment, cells were stained with 5 µM MitoPerOx reagent for 30 minutes at 37 °C in the dark. Following incubation, the cells were washed twice with PBS. Fluorescence was measured using a multimode microplate reader (Infinite 200 Pro, Tecan, Switzerland) with excitation at 495 nm and dual emission measurements at 520 nm and 582 nm. The ratio of fluorescence intensity (582 nm/520 nm) was calculated to indicate mitochondrial lipid peroxidation.
Flow Cytometric Analysis of MitoSOX RedMitochondrial superoxide levels were measured using MitoSOX Red (M36005, Thermo Fisher Scientific, USA). Briefly, cells were seeded at a density of 2 × 105 per well in 12-well plates and treated under specified conditions. After treatment, cells were washed with PBS and incubated with 5 µM MitoSOX Red reagent in serum-free medium at 37 °C for 30 minutes in the dark. Subsequently, the cells were harvested, washed twice with PBS, and resuspended in 500 µL of PBS. Mitochondrial superoxide was quantified using a BD LSRFortessa flow cytometer (BD Biosciences, USA), equipped with a 488 nm excitation laser, and fluorescence emission was detected in the PE channel (575/26 nm). Data were analyzed using FlowJo v10.8.
JC-1 StainingMitochondrial membrane potential was assessed using a JC-1 Assay Kit (C2006, Beyotime, China) according to the manufacturer’s instructions. Cells were seeded in Thermo Scientific™ Nunc MicroWell 96-well plates (165305, Thermo Fisher Scientific, USA) with clear bottoms at a density of 1 × 104 cells per well and treated accordingly. After treatment, cells were incubated with JC-1 working solution (5 µg/mL) for 30 minutes at 37 °C in the dark. Following incubation, the supernatant was gently removed, and cells were washed twice with pre-warmed JC-1 staining buffer. Fluorescence was measured using a multimode microplate reader (Infinite 200 Pro, Tecan, Switzerland). The JC-1 aggregates (polarized mitochondria) were detected with an excitation wavelength of 525 nm and an emission wavelength of 590 nm (red fluorescence), while JC-1 monomers (depolarized mitochondria) were detected with an excitation of 490 nm and an emission of 530 nm (green fluorescence). The ratio of red (aggregate) to green (monomer) fluorescence intensity was calculated to evaluate mitochondrial depolarization.
Assessment of Apoptosis by Annexin V-FITC/PI StainingApoptosis was assessed using the Annexin V-FITC/PI Apoptosis Detection Kit (K2003, APExBIO Technology, USA). Cells were seeded in 12-well plates at a density of 1 × 105 cells per well and treated under specified conditions. After treatment, cells were collected, washed with cold PBS, and resuspended in 195 µL of 1× binding buffer. Then, 5 µL of Annexin V-FITC and 5 µL of propidium iodide (PI) were added and incubated for 15 minutes at room temperature in the dark. Samples were analyzed within 1 hour using a BD LSRFortessa flow cytometer (BD Biosciences, USA). FITC fluorescence was detected using the FITC channel (excitation 488 nm, emission 530/30 nm), and PI was measured in the PE channel (excitation 488 nm, emission 585/42 nm). Data were analyzed using FlowJo v10.8. The percentage of apoptotic cells was defined as the population positive for Annexin V-FITC.
Exosome Uptake in SH-SY5Y Neuronal CellsExosome uptake was assessed using both flow cytometry and fluorescence microscopy. SH‑SY5Y cells were seeded onto glass coverslips in 48‑well plates (2 × 104 cells/well) and serum‑starved for 12 hours. Plasma‑derived exosomes were labeled with 5 µM Dil (C1036, Beyotime, China) for 20 minutes at 37 °C. Unbound dye was removed using a spin‑column‑based exosome dye removal kit (E0015, Mele biotech, China). Cells were treated with Dil‑labeled exosomes at concentrations of 10, 20, 30, or 50 µg/mL for 24 hours. A dye‑only control was prepared by subjecting Dil in PBS to the same purification steps in the absence of exosomes.
For fluorescence microscopy, cells were fixed with 4% paraformaldehyde for 15 minutes, permeabilized with 0.3% Triton X‑100 (X100, Sigma-Aldrich, USA) for 10 minutes, and blocked with 5% BSA (A8806, Sigma-Aldrich, USA) for 1 hour. They were then incubated overnight at 4 °C with an anti‑β‑III‑tubulin (Tuj1) antibody (ab18207, rabbit, 1:200, Abcam, USA), followed by incubation with an Alexa Fluor 488‑conjugated secondary antibody (A‑11008, 1:1000, Invitrogen, USA) for 1 hour. Nuclei were stained with DAPI (62248, Thermo Fisher Scientific, USA). Images were acquired using a confocal microscope (Olympus FV3000); Z‑stack images were collected at 0.54 μm intervals. Three‑dimensional reconstruction, surface rendering (Tuj1, green), and quantification of intracellular exosome puncta (Dil, red) were performed using IMARIS 10.0.1 (Bitplane Imaris, Oxford, UK). Co‑localization analysis confirmed the intracellular localization of the Dil signal.
For flow cytometric quantification, after 24 hours of treatment, cells were washed twice with cold PBS, detached with trypsin‑EDTA, and collected by centrifugation. Cell pellets were resuspended in 500 µL of PBS and passed through a cell strainer to obtain single‑cell suspensions. Fluorescence intensity of Dil was measured (excitation 488 nm, emission 575/26 nm, PE channel) using a BD LSRFortessa flow cytometer (BD Biosciences, USA). Data were analyzed with FlowJo software (v10.8), and the geometric mean fluorescence intensity (MFI) in the PE channel was calculated for each condition to quantify exosome uptake.
Statistical AnalysisData are expressed as mean ± standard error of the mean (SEM). Normality was assessed using the Shapiro–Wilk test. For normally distributed data, comparisons between two groups used an unpaired Student’s t-test, while comparisons among three or more groups used one-way ANOVA with Tukey’s post hoc test (assuming equal variance, confirmed by Brown–Forsythe test). Non-normally distributed data were analyzed with the Mann–Whitney U test (two groups) or Kruskal–Wallis’s test with Dunn’s post hoc test (multiple groups). Outliers were identified and excluded using the ROUT method (Q = 1%). Statistical significance was set at p-value < 0.05 (two-tailed). Analyses were performed using GraphPad Prism 9.0.
Comments (0)