
Remember me
This multicenter, prospective, observational study will enroll 720 patients with DPNP treated with mirogabalin across 36 sites in China. Participating centers will include hospitals with established expertise in managing DPNP. Sites will be selected to ensure geographic diversity and the capacity to maintain patient follow-up throughout the study period, thereby supporting the enrollment of a nationally representative patient sample.
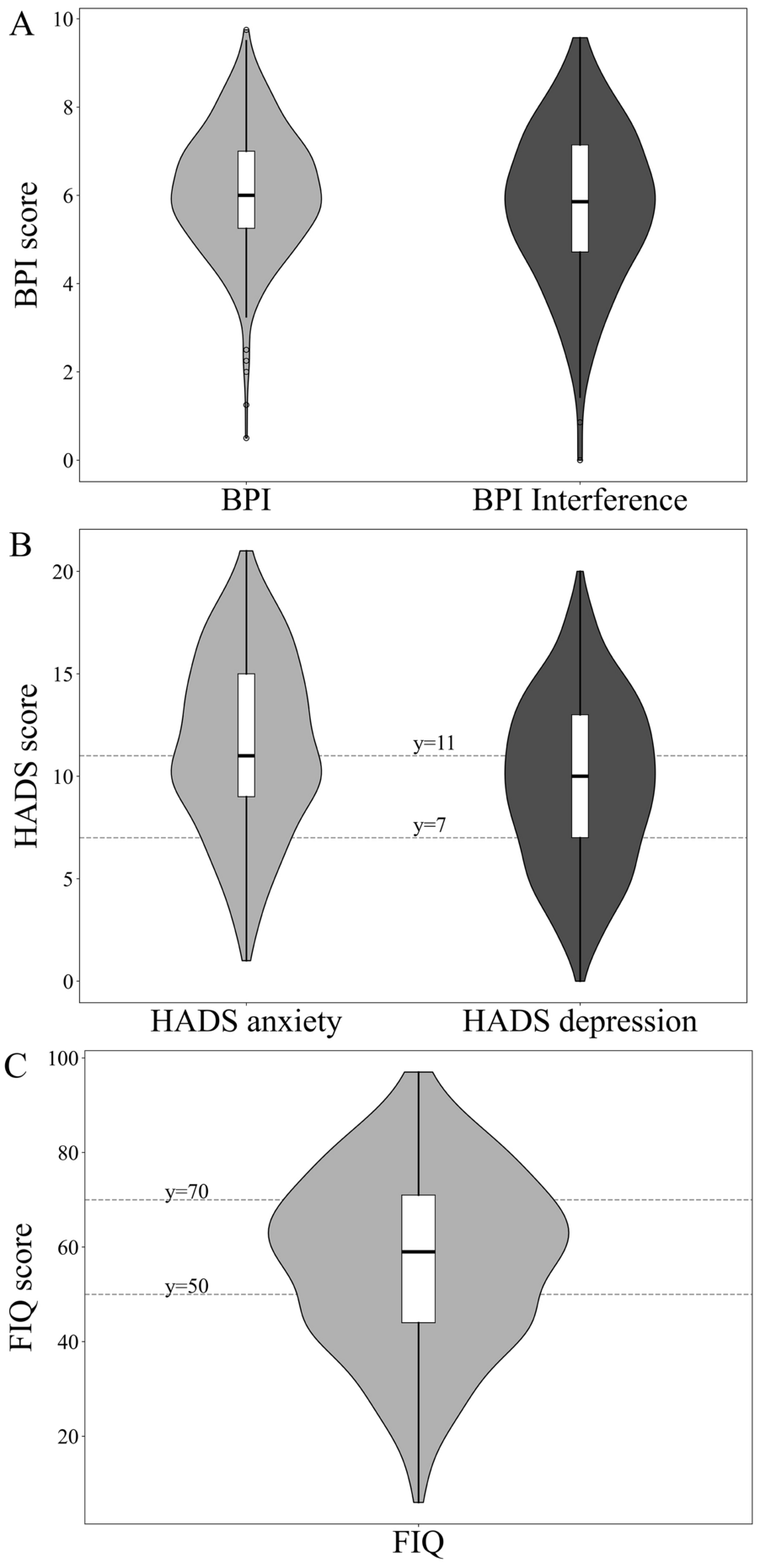
Patients diagnosed with DPNP and who are treatment-naïve will be eligible for consecutive enrollment in this study. Baseline data will be collected within 30 days prior to, or on the date of, obtaining written informed consent (ICF) from the patients. The follow-up period is defined as the duration from the ICF until the earliest occurrence of one of the following: completion of the 15-week observation period (including a 14-week mirogabalin treatment phase and one additional week), completion of a 1-week observation period following early discontinuation of mirogabalin (if treatment duration is < 14 weeks), withdrawal of consent, loss to follow-up, or study termination. Patient outcomes will be assessed throughout this period as outlined in the study design (Fig. 1). Due to the observational nature of this study, all follow-up visits and procedures will depend on routine practice, with no scheduled time points as per protocol. The time window of follow-up measurement is defined on the basis of patients taking mirogabalin as day 1, and week 1–14 were set to the duration of (1–7)* number of weeks. The minimum assessment frequency is once per week for visual analog scale (VAS) score and once per visit for douleur neuropathique 4 questionnaire (DN4) score. In cases of participant withdrawal, standard medical care and treatment will be continued as per routine clinical practice; observations up to the final dose of mirogabalin will be documented, and reasons for early discontinuation will be recorded in the electronic case report form (eCRF).
Fig. 1 The alt text for this image may have been generated using AI.
The alt text for this image may have been generated using AI.Study design of ReMIssion. Data collection: available data will be collected; participants’ diagnosis, treatment, and monitoring procedures will follow routine clinical practice. ePRO electronic patient-reported outcome, ICF informed consent form, V visit
Patient PopulationEligible patients include adults aged ≥ 18 years at the time of signing the ICF, those diagnosed with DPNP, those with a VAS pain score of ≥ 40 mm on the day the ICF is signed, those who are mirogabalin-naïve before signing the ICF and planning to initiate treatment with mirogabalin, those who signed the ICF for study participation, and those willing to provide access to previous and future medical information.
Patients with severe pain unrelated to DPNP at baseline, with a history of gabapentin therapy (≥ 1200 mg/day) or pregabalin (≥ 300 mg/day) for DPNP that was ineffective, received crisugabalin or any other analgesic medications, including TCAs (amitriptyline and nortriptyline), SNRIs (duloxetine and venlafaxine), antiepileptics (carbamazepine), and opioids (tapentadol and tramadol), within 7 days prior to ICF is signed, had major psychiatric disorders at baseline, or with contraindications for mirogabalin treatment as per China’s prescribing information for mirogabalin will be excluded from the study. The 7-day washout period was chosen on the basis of the pharmacokinetic properties of the analgesics listed in the exclusion criteria and can minimize residual effects on baseline pain assessments.
Planned OutcomesThe primary outcome is the proportion of participants with any treatment-related adverse events (TRAEs). A TRAE is defined as having a reasonable temporal sequence from drug administration and that cannot be reasonably explained by the participant’s clinical state or other factors (e.g., disease under study, concurrent diseases, concomitant medications) or a known reaction to the drug or its chemical group or predicted by known pharmacology. Investigators will review the cases of adverse events (AEs) and assess causality between drug and AE on the basis of evaluation standard and available adverse events information. Secondary outcomes are time-to-onset and symptom duration of somnolence, dizziness, weight gain, and peripheral edema; the proportion of patients with treatment-emergent adverse events (TEAEs), serious TEAEs, and TRAEs leading to study treatment discontinuation; the change from baseline in the VAS score at week 14; the proportion of participants achieving 30% and 50% pain relief at weeks 4, 8, and 14; time to achieve 30% or 50% pain relief; the change from baseline in the DN4 score at week 14; and the patient global impression of change (PGIC) and change from baseline in the EuroQol Five-Dimension Five-Level (EQ-5D-5L) score at week 14.
Exploratory outcomes are dosage and dosing frequency of mirogabalin, including the initial daily dose, maximum daily dose, maintenance dose, time to reach maximum daily dose, duration of maintenance at maximum daily dose, adherence to mirogabalin measured by proportion of days covered (PDC), and the level of treatment satisfaction at week 14 or at the end of mirogabalin treatment.
ExposureAs this is a noninterventional observational study, all enrolled patients will receive diagnosis and treatment according to routine clinical practice. No investigational product will be evaluated. The decision to initiate mirogabalin treatment will be made independently of study participation, on the basis of solely the treating physician’s clinical judgment in consultation with the patient, as part of standard care.
Details related to mirogabalin exposure (e.g., treatment start and end dates, dosage, frequency, and reasons for any changes or discontinuation) will be collected retrospectively from patients’ medical records and documented in the eCRF.
Data CollectionBaseline data will include demographics at the time of enrollment, medical and treatment history, vital signs, and adverse events (AEs)/serious AEs (Table 1). All data, except for patient-reported outcomes (PROs), will be collected from medical records after the ICF is signed via a customized eCRF, complemented with investigators’ evaluations at baseline and thereafter at follow-up visits according to clinical practice until the end of the study. PRO data, including VAS, patient global impression of change (PGIC), EQ-5D-5L, and treatment satisfaction questionnaire, will be collected via electronic patient-reported outcome (ePRO) at different time points (Table 2). VAS will be assessed at baseline, at least four times during week 1, three times during week 2, two times during week 3, and at least one time a week for the rest of the treatment period until week 14 or the end of mirogabalin treatment. PGIC and mirogabalin treatment-related satisfaction questionnaire will be assessed at week 14 or at the end of treatment. EQ5D-5L will be assessed at baseline and at week 14 or at the end of treatment (Table 2). Given the observational design, no mandatory visits or protocol-driven procedures are required; only available and identifiable assessments will be recorded, with data collection intervals set at weeks 4, 8, 12, and 15 to serve as reminders for routine data collection.
Table 1 Data collected from medical recordsTable 2 Assessment of patient-reported outcomesQuality Control and Quality AssuranceThis study will be conducted in accordance with the principles of good pharmacoepidemiology practices (GPP), good pharmacovigilance practices (GVP) and good clinical practice (GCP). A contract research organization (CRO) will be responsible for data management activities, including data entry oversight and validation. Data will be collected using an electronic data capture (EDC) system, with study sites. The trained investigators from study sites are responsible for entering the data and ensuring the accuracy and completeness of all collected data.
Appropriate quality control measures, including data plausibility checks and routine data monitoring, will be implemented throughout the study to ensure the integrity and reliability of the data. Ongoing data quality checks will be performed regularly, and any queries generated will be addressed by the respective sites throughout the study conducting. On-site monitoring will be conducted as per the Clinical Monitoring Plan, with verification of informed consent documentation and source data against the participants’ medical records (100% for key source data and approximately 30% for total amount of data). The data management plan (DMP) will be used during the study. Data review (100% of total data) will be performed by data manager per DMP during the study conducting period to ensure data quality. Consistency verification of external data, medical coding, and so on will also be performed. The medical monitoring plan will be prepared and medical monitoring will be started when ten patients are enrolled and performed regularly when 5%, 10%, 20%, 30%, 40%, 50%, 60%, 80%, and 100% of patients are enrolled with the maximum 2 months of every data output to ensure medical consistency and avoid the potential underreporting of AEs. A safety-related follow-up will be made to obtain more information of AEs. On the basis of new or confirmed information, investigators will also perform re-evaluation to make assessment more scientific and comprehensive.
Sample Size EstimationThe primary objective of the ReMIssion study is to assess the safety of mirogabalin treatment in patients with DPNP in routine clinical practice in China. All analyses will be descriptive and explorative, and no formal hypothesis will be tested. Hence, sample size calculations are based on precision.
In a phase 3 study involving Chinese patients, the incidence of TEAEs, dizziness, somnolence, weight gain, and peripheral edema in patients treated with mirogabalin was 6.6%, 6.1%, 5.6%, and 5.1%, respectively [8]. If the proportion of patients with any adverse event (AE) is assumed to be 50%, 720 evaluable patients will achieve a precision (or half-width of the confidence intervals [CIs]) of 3.65%, estimated by the Wald method. If the proportion of patients with any AE is assumed to be 1%, 720 evaluable participants will achieve a precision (or half-width of CIs) of 0.73%, estimated by the Wald method. Accordingly, the 720 participants will be sufficient to have a 94.4% probability of observing at least one case if the proportion of participants with any AE is ≥ 0.4%.
Statistical AnalysesOwing to the descriptive and explorative nature of the ReMIssion study, no formal hypothesis will be tested. Descriptive statistics will be used for all analyses. Statistical tests will be performed as appropriate. All statistical tests will be two-sided, with P-values < 0.05 considered as statistically significant. Nominal P-values will be presented only for exploratory purposes. The safety and effectiveness analysis sets will include all patients who received at least 1 dose of mirogabalin.
For the safety analyses, all TRAEs and TRAEs leading to discontinuation of the study treatment will be summarized. In addition, major TRAEs will be reported for patients’ subgroups with different degrees of renal impairment, participants with different severities of pain, and participants who are treatment-naïve or have received prior treatment for DPNP.
For the effectiveness analyses, continuous variables will be summarized by the number of observations, with mean (standard deviation [SD]), or median (interquartile range). Categorical variables will be summarized using frequency and percentage. PRO data will be analyzed using proper methods, corresponding to the outcomes under evaluation and the research questions associated with each study objective. Effectiveness will be assessed by summarizing the changes in VAS scores from baseline at each time point, including descriptive summaries at week 14. Mixed models for repeated measures (MMRM) will be performed to assess the changes in VAS scores from baseline to each time point with time points as the fixed effect and baseline VAS measurement as a covariate. Responder rates (≥ 30% and ≥ 50% pain reduction) at weeks 4, 8, and 14 will be reported with 95% CIs. Time to response will be analyzed using the Kaplan–Meier estimates. DN4 score changes from baseline at week 14, PGIC scores, and EQ-5D-5L outcomes will also be summarized descriptively. Exploratory analyses will involve descriptive statistics to summarize the real-world usage patterns of mirogabalin and patient satisfaction levels among Chinese patients with DPNP.
Methods to Minimize BiasObservational studies based on data derived from routine clinical practice are inherently susceptible to biases, including selection bias, information bias, and confounding. To mitigate selection bias, site selection will take into account geographic distribution to ensure a more nationally representative sample of patients, physicians, and clinical practices. Investigators will be encouraged to consecutively enroll all eligible patients to support unbiased recruitment. To reduce information bias, participating sites will be instructed to enter both retrospective and prospective study data as completely and promptly as possible. For any missing data, descriptive analyses will be performed to summarize the missing rate and the pattern of missing variables. No imputation will be performed for missing safety data. Missing data for effectiveness endpoints will be imputed using multiple imputation methods or other approaches such as last observation carried forward (LOCF), as appropriate, according to the definition of the estimand. In addition, subgroup analysis of outcomes (such as VAS, time to response), supportive analysis, or sensitivity analysis will be performed as appropriate.
Ethical ConsiderationsThe ReMIssion study protocol, amendments, ICFs, and information sheets will be approved by the relevant independent ethics committee (IEC) or institutional review board (IRB) of each study center. The study will be conducted in compliance with the principles of the Declaration of Helsinki and the International Council for Harmonisation (ICH) consolidated guideline E6 for good clinical practice. All participants will provide written informed consent prior to study participation. The ReMIssion study is registered with ClinicalTrials.gov under the identifier NCT06812117.
Comments (0)