Study Design
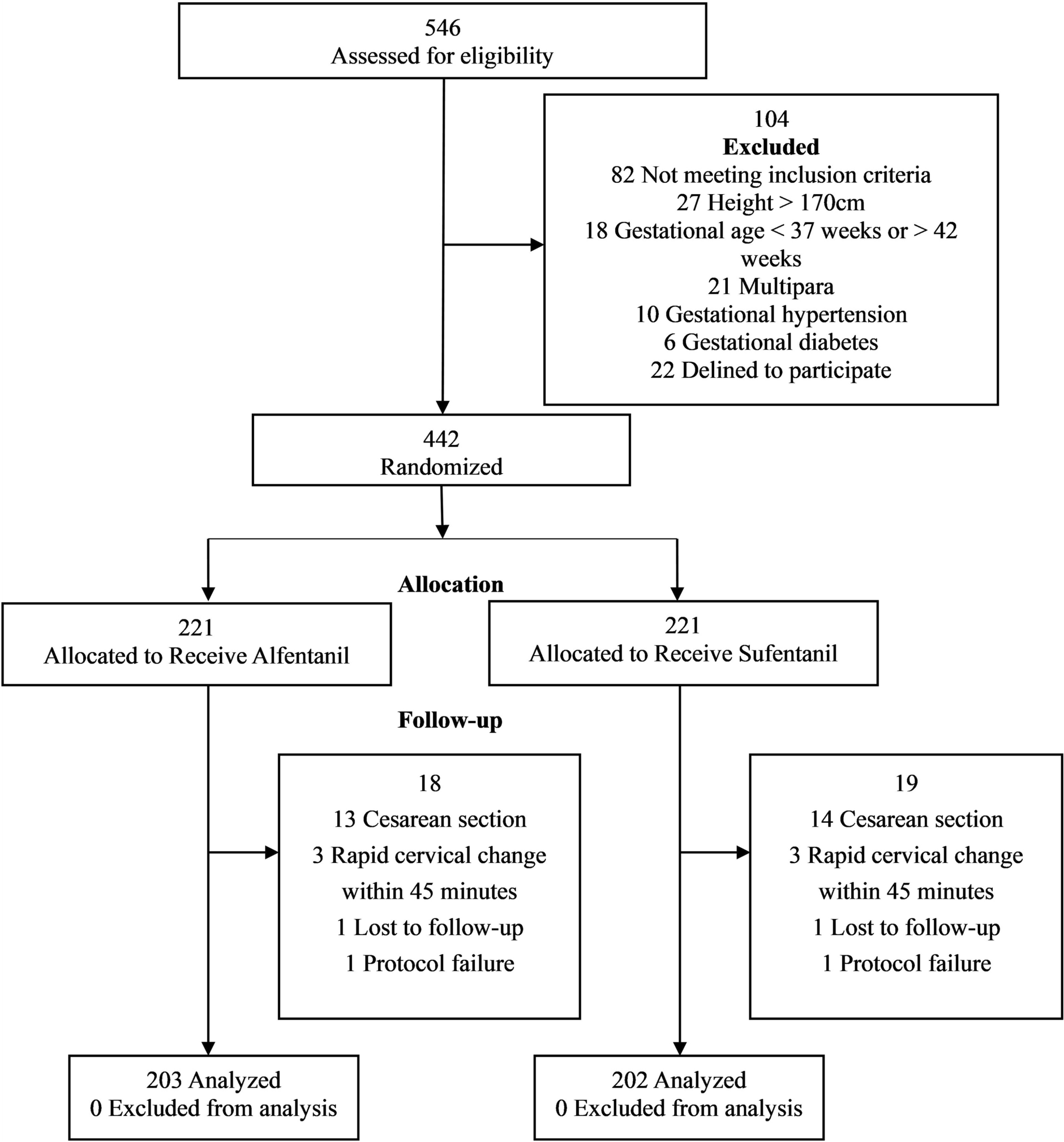
This was a prospective, randomized, controlled, single-blind trial with two parallel groups, conducted at four tertiary hospitals in China. Six hospitals were registered, but only four completed the study. The study protocol was approved by the Ethics Committee of the General Hospital of the Northern Theater Command of the Chinese People’s Liberation Army (Approval No. Y [2023] 069). All participating sites accepted the central approval. The clinical trial registration number was ChiCTR2300072104, and the period of the study was from June 2023 to June 2024. The study protocol was conducted in accordance with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Written informed consent was obtained from all participants prior to enrollment. This study complies with the ICMJE recommendations on clinical trial registration and transparency of results reporting [14].
Before initiation, all investigators from the participating centers received standardized training from the principal investigator. The training covered clinical procedures, outcome assessment, and data collection to ensure consistency and objectivity across sites.
Participants
Eligible parturients were nulliparous women with full-term singleton pregnancies (37–42 weeks) in vertex presentation. They were required to be 20–35 years of age, with an American Society of Anesthesiologists (ASA) physical status of II, height between 150 and 170 cm, and body mass index (BMI) between 20 and 35 kg/m2. Enrollment occurred when cervical dilation reached 2–3 cm with regular uterine contractions, and suitability for vaginal delivery was confirmed by the attending obstetrician and midwife.
Women were excluded if they had contraindications to neuraxial anesthesia, including coagulopathy, infection at the puncture site, uncorrected hypovolemia or hypotension, elevated intracranial pressure, or severe spinal deformities. Pregnancies complicated by gestational diabetes, preeclampsia, or placenta previa, as well as significant maternal cardiac, neurologic, hepatic, or renal disease, were also excluded. Fetuses with intrauterine growth restriction or congenital malformations were not eligible.
Additional exclusion criteria included inability to cooperate with epidural placement or pain scoring, a baseline visual analogue scale (VAS) score < 6 before analgesia, allergy to local anesthetics or opioids, or prior opioid administration. Participants were withdrawn if epidural catheterization failed or accidental dural puncture occurred, if severe adverse events developed such as profound hypotension, local anesthetic systemic toxicity, or total spinal anesthesia, if conversion to cesarean section became necessary, if informed consent was withdrawn, or if rapid cervical dilation (≥ 5 cm within 45 min of epidural initiation) was observed.
Group Allocation
Participants were randomly assigned in a 1:1 ratio to either the sufentanil group (group S) or the alfentanil group (group A) using a computer-generated random number table. Allocation was concealed until the initiation of epidural analgesia, and parturients were blinded to group assignment.
The choice of study doses was based on prior pharmacological evidence. Under the PIEB regimen with 0.075% ropivacaine, the 90% effective concentration (EC90) of alfentanil was reported as 3.85 μg/ml (95% CI, 3.65–4.28 μg/ml) [12]. Considering the analgesic potency ratio of alfentanil to morphine (40–50:1) and sufentanil to morphine (approximately 1000:1), the equipotency ratio between alfentanil and sufentanil was estimated to be 20–25:1.
Based on these calculations, the final study solutions were defined as 0.075% ropivacaine combined with 0.2 μg/ml sufentanil in group S and 0.075% ropivacaine combined with 4 μg/ml alfentanil in group A.
Randomization and Blinding
A center-stratified block randomization scheme with randomly varying block sizes (4 or 6) was employed to ensure balance within each center. An independent statistician generated the allocation sequence with SAS 9.4. “Center” served as the stratification factor (four strata).
Within each stratum, participants were randomly assigned to either group S or group A in a 1:1 ratio using permuted blocks of length 4 or 6. The 442 unique randomization numbers (ID 001–ID 442) were pre-distributed across centers according to anticipated recruitment.
The sequence was uploaded to a central web-based randomization system (IWRS) that was inaccessible to investigators before randomization. After confirming eligibility, the investigator logged into the system, entered the participant’s details, and immediately received the next randomization number and corresponding group assignment, thereby maintaining allocation concealment. Because the trial was single-blind, only participants were blinded to group allocation; investigators were aware of the assigned treatment and responsible for its administration and follow-up. The statistician who performed the primary analyses remained blinded to group membership until the database was locked. This was a single-blind trial: only the participants were blinded to group assignment, whereas drug preparation, dispensing, monitoring, and all clinical management were conducted by an unblinded study team.
Epidural Analgesia Procedure
Upon admission to the labor ward at 2–3-cm cervical dilation, routine monitoring was established, including electrocardiogram, noninvasive blood pressure, heart rate, saturation of peripheral oxygen, uterine contractions, and fetal heart rate. An intravenous line was secured, and lactated Ringer’s solution was administered.
With the patient in the left lateral position, epidural puncture was performed at the L2–3 or L3–4 interspace (preferably L2–3). The epidural catheter was advanced 4 cm cephalad. After confirming the absence of cerebrospinal fluid or blood aspiration, a test dose of 3 ml 1% lidocaine was injected. Upon negative test results, the catheter was fixed in place. Patients with dural puncture or failed catheterization were excluded.
The catheter was connected to an electronic analgesia pump programmed for PIEB combined with PCEA. The regimen was: PIEB bolus dose 10 ml, interval 50 min, background infusion 3 ml/h; PCEA bolus 8 ml, lockout time 20 min. Effective analgesia was defined as VAS score ≤ 3 within 30 min after epidural initiation.
A VAS score above 3 at 30 min after epidural labor analgesia was considered inadequate analgesia. In cases of incomplete analgesia, the possible causes were considered and appropriate measures were taken according to the manifestations of incomplete analgesia. For example, if the analgesic plane was adequate (T10-S4) but the analgesic intensity was insufficient, the concentration of analgesic could be increased. For bilateral blocks and insufficient analgesic planes, a large volume (5–15 ml) of low-concentration local anesthetic was used for planar diffusion.
Outcomes
The selection of outcome measures was guided by the Core Outcome Measures in Effectiveness Trials (COMET) Initiative recommendations for developing core outcome sets [15]. The primary outcome of this study was the onset of analgesia, defined as the median time from drug administration to the first recording of a VAS score ≤ 3 within 30 min. The VAS score was evaluated every 2 min within 30 min of epidural analgesia.
Secondary outcomes encompassed several maternal parameters, including hemodynamic variables (blood pressure, heart rate, and oxygen saturation), intrauterine pressure, temperature and fetal heart rate, which were recorded at baseline, 15, 30, and 45 min after initiation of epidural analgesia, and again at full cervical dilation. Pain intensity was assessed using the VAS, while motor block was evaluated with the Bromage scale. The highest level of sensory block and maternal body temperature were also documented.
Additional indicators included the duration of analgesia, length of labor, mode of delivery, oxytocin requirement, and total anesthetic consumption (total volume of PIEB solutions and total doses of opioids used). Neonatal condition was assessed by Apgar scores at 1 and 5 min after birth. Maternal satisfaction was rated on a 0–10 scale, and adverse events such as pruritus, nausea, vomiting, hypotension, urinary retention, and lower limb numbness were systematically recorded. Secondary outcomes that were not pre-specified in the trial registry were analyzed post hoc, including intrauterine pressure, maternal temperature, maternal satisfaction scores, neonatal Apgar scores, and oxytocin requirement. However, all data were routinely captured in pre-specified case report form (CRF) fields as part of standard obstetric monitoring and required no additional measurements or retrospective data extraction.
Sample Size Calculation
Based on pilot data, the mean onset time was 8.1 ± 1.6 min with 0.2 μg/ml sufentanil + 0.075% ropivacaine and 6.6 ± 1.4 min with 4 μg/ml alfentanil + 0.075% ropivacaine. With α = 0.05 and β = 0.10 (power = 90%), a sample size of 201 participants per group was required. Assuming a 10% dropout rate, 221 parturients were planned per group, yielding a total of 442 participants.
Statistical Analysis
The primary analysis was conducted on the per-protocol (PP) population, including only those participants who completed the intervention as assigned without major protocol violations, to assess the consistency of the findings. Data were analyzed using SPSS version 27.0. Continuous variables with normal distribution were expressed as mean ± standard deviation (SD) and compared using independent-samples t tests. Non-normally distributed data were expressed as median (interquartile range, IQR) and compared with the Mann–Whitney U test. Median differences (and 95% CIs) were calculated with Hodges–Lehmann estimators. Categorical data were expressed as counts (percentages) and compared using chi-square tests. Repeated measurements across time points were analyzed using generalized estimating equations (GEE), including time-by-group interactions. A two-sided P < 0.05 was considered statistically significant.

Comments (0)