Ethical Approval
This was a single-center, randomized, double-blind, controlled trial. The study protocol, including the use of a saline-control ICNB group, was reviewed and approved by the Institutional Review Board (IRB) of Foshan Hospital of Traditional Chinese Medicine (approval no. KY[2023]153). The IRB approved the use of a saline placebo after determining that the scientific value of having a rigorous control group outweighed the minimal risks associated with the ultrasound-guided procedure when performed by experienced clinicians. The trial was retrospectively registered with the Chinese Clinical Trial Registry (www.chictr.org.cn, number: ChiCTR2500105786). The delay between ethical approval and trial registration was an administrative oversight. We emphasize that all procedures, including patient recruitment and data collection, were initiated only after obtaining ethical approval from the IRB. The study was conducted in full adherence to the approved protocol and the principles of the Declaration of Helsinki and the CIOMS guidelines. Written informed consent was obtained from all patients. The consent form explicitly stated that patients might receive an ultrasound-guided injection of normal saline as part of the study group allocation, clarifying that this substance lacks an anesthetic effect.
Study Participants
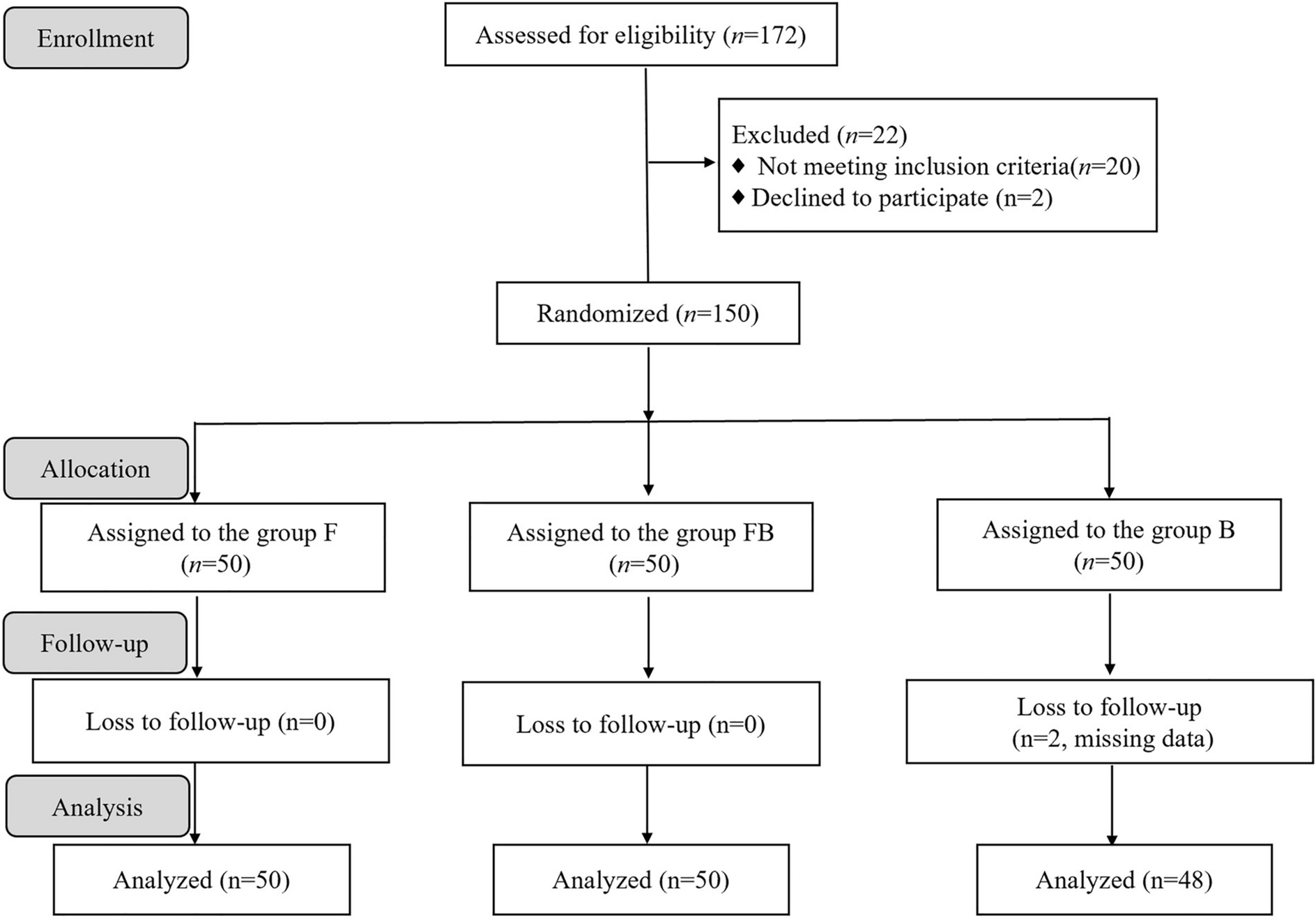
Adult patients (aged ≥ 18 years) diagnosed with traumatic multiple rib fractures (the rib fracture pain was the patient's dominant source of pain) at or after admission and scheduled for surgical treatment were assessed for eligibility. Exclusion criteria included American Society of Anesthesiologists (ASA) physical status IV or higher, body weight < 50 kg, body mass index (BMI) > 35 kg/m2, presence of flail chest or paradoxical respiration, inability to clearly identify rib fracture pain as the patient's dominant source of pain (e.g., significant long bone fractures, visceral injuries, or traumatic brain injury), requirement for intubation and mechanical ventilation, known allergy to flurbiprofen axetil or tramadol or ropivacaine, and inability to comprehend or utilize the Visual Analog Scale (VAS) for pain assessment. The exclusion of patients with flail chest was a deliberate choice to enhance the homogeneity of the study population. Flail chest is associated with more profound pulmonary dysfunction, a higher incidence of lung contusions, and a greater likelihood of requiring mechanical ventilation. These factors could significantly confound the assessment of analgesic efficacy, particularly for respiratory outcomes [14]. Furthermore, the pain profile and analgesic requirements in flail chest are often qualitatively different and more complex, typically necessitating more intensive or alternative analgesic strategies (e.g., continuous epidural infusion) that would not be comparable to the standardized regimens under investigation in this trial [13]. This study therefore focused on a distinct group of surgical patients with multiple rib fractures in the absence of flail chest. Patients were enrolled from March to November 2024.
Randomization, Grouping, and Blinding
A computer-generated randomization sequence was created by an independent statistician using SPSS software (version 26.0), allocating patients in a 1:1:1 ratio to one of three parallel groups. The allocation sequence was concealed using sequentially numbered, opaque, sealed envelopes, which were stored securely and opened sequentially only after a patient had provided written informed consent and been assigned a study identification number. Patients were randomized into three groups: Group F received intravenous flurbiprofen axetil (50 mg) plus an ultrasound-guided ICNB with normal saline; Group FB received intravenous flurbiprofen axetil (50 mg) plus an ultrasound-guided ICNB with 0.4% ropivacaine; Group B received intravenous normal saline plus an ultrasound-guided ICNB with 0.4% ropivacaine.
This study employed a double-blind design. The patients, anesthesiologists performing the outcome assessments, and data analysts were all blinded to group assignment. The unblinded anesthesiologist (Anesthesiologist A), who was not involved in patient recruitment, clinical procedures, or data collection, prepared all study drugs according to the randomization schedule. To ensure blinding, all syringes were wrapped in opaque covers to mask their contents, and the unblinded anesthesiologist had no further contact with the patients or the research team after preparing the medications. The interventions were administered by a separate blinded anesthesiologist (Anesthesiologist B).
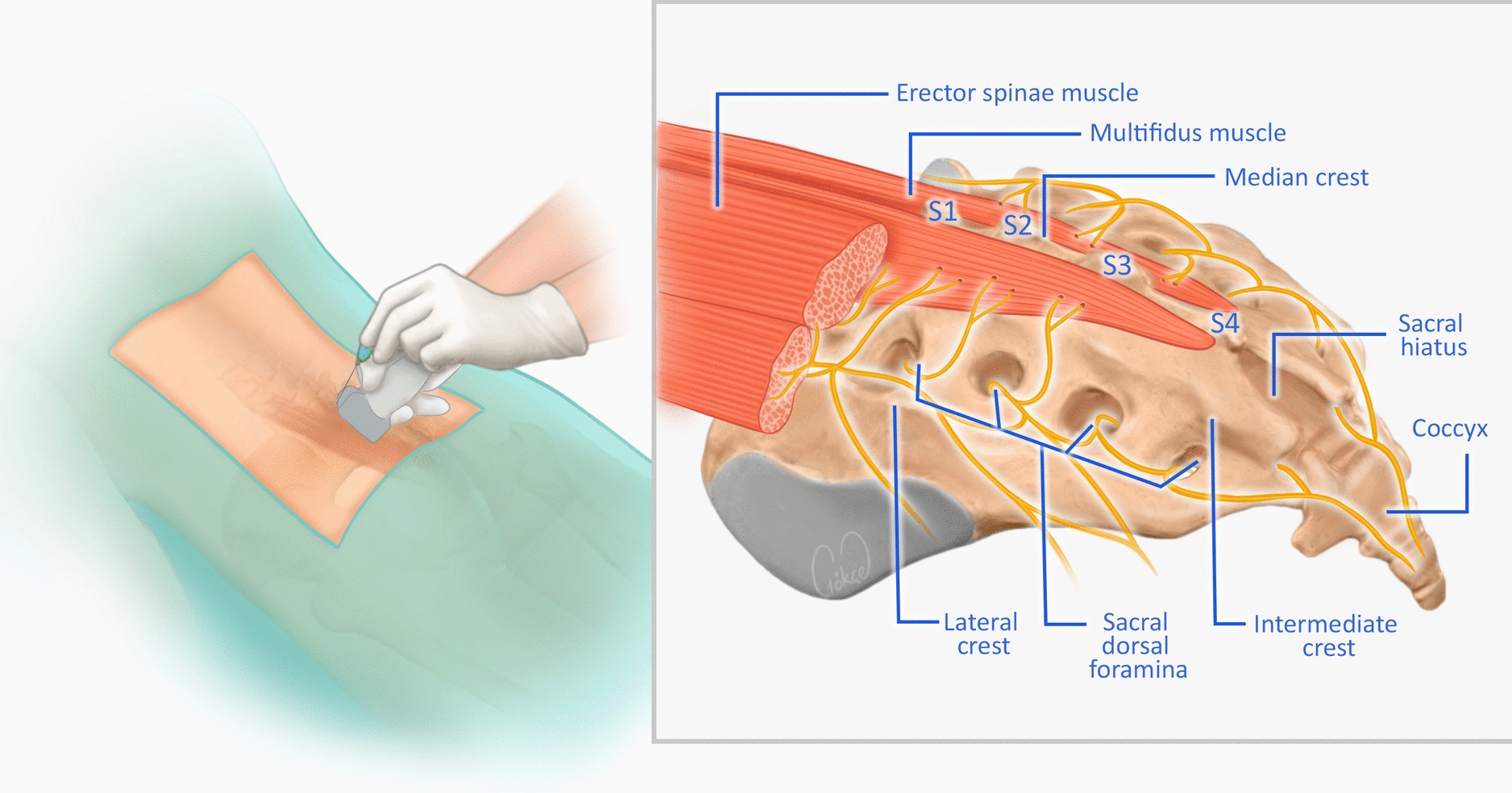
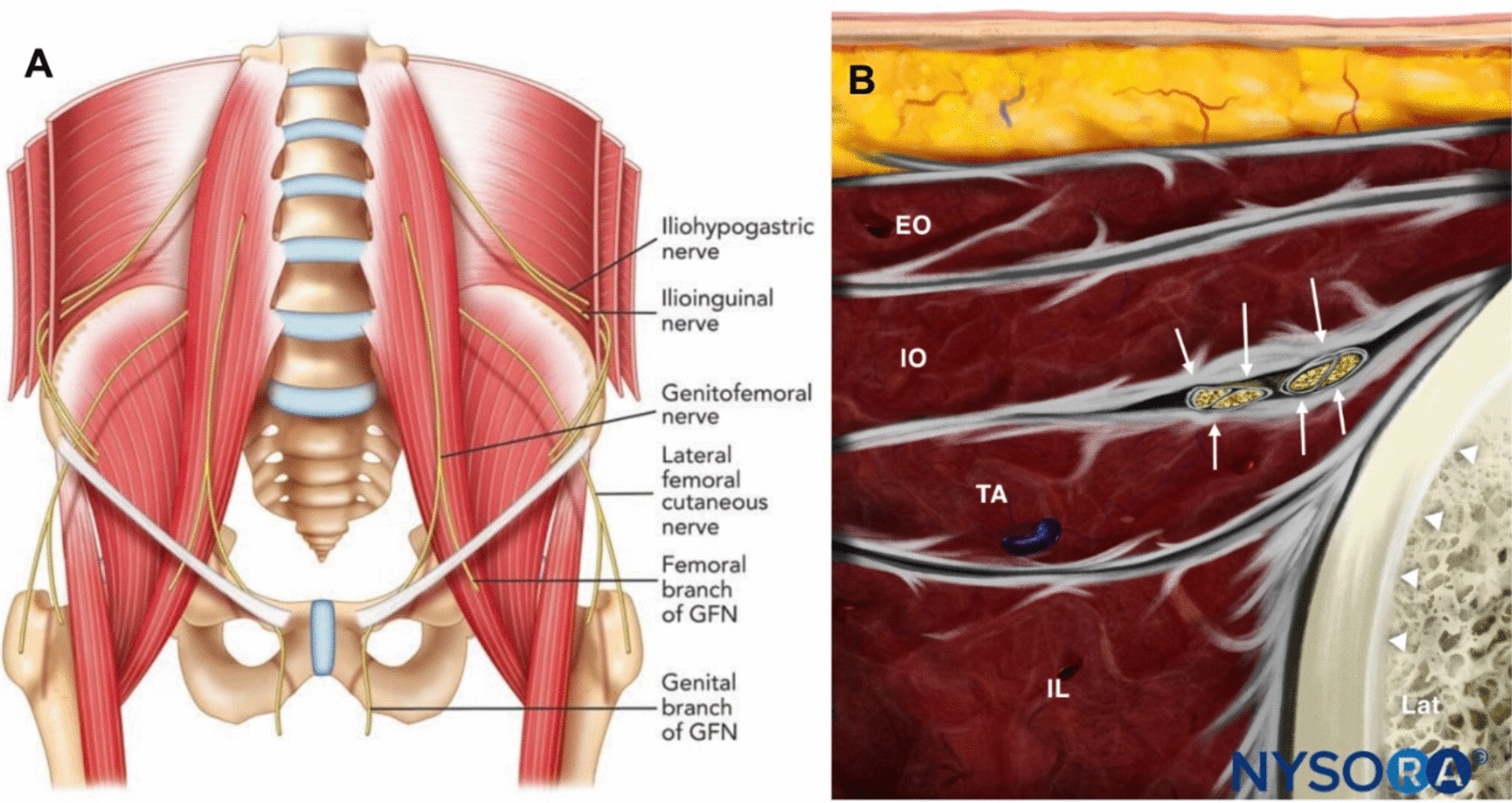
Ultrasound-Guided ICNB and Diaphragmatic Excursion Procedure
Before the procedure, imaging studies were reviewed to identify the fractured rib segments.
The patient was placed in the lateral decubitus position with the side scheduled for nerve block facing upward. The 12th rib was identified and marked, and the probe was then moved cephalad, tracking and marking the ribs from the 11th up to the 1st. After standard aseptic preparation, a 21-gauge needle was advanced using an in-plane technique toward the intercostal space at the rib angles. The needle tip was positioned between the internal and innermost intercostal muscles. Following negative aspiration for blood or air, 2 ml of the study solution (0.4% ropivacaine or normal saline) was injected per nerve, creating a visible hypoechoic fluid spread. The block was performed at all levels corresponding to the fractured ribs.
Diaphragmatic excursion was measured using the same ultrasound machine equipped with a curvilinear probe, following previously described methodologies in the literature [15, 16].
Arterial Blood Gas Sampling and Analysis
Arterial blood samples for gas analysis were collected via radial artery puncture under aseptic conditions 30 min after the completion of the intercostal nerve block. Throughout the preoperative period, including at the time of blood sampling, patients breathed room air (FiO2 0.21) unless their peripheral oxygen saturation (SpO2) was < 90%, in which case supplemental oxygen was administered via nasal cannula or face mask at a low flow rate (2–5 l/min) to maintain SpO2 ≥ 90%. A 22-gauge needle and a heparinized syringe were used to draw approximately 2 ml of arterial blood. The samples were immediately placed on ice and analyzed within 10 min of collection using a portable blood gas analyzer (model: RAPIDPOINT 500, manufacturer: Siemens Healthcare Diagnostics Inc.) to measure the pH, partial pressure of oxygen (PaO2), partial pressure of carbon dioxide (PaCO2), SpO2, and calculation of the oxygenation index (PaO2/FiO2). The FiO2 value was recorded at the time of sampling to ensure accurate calculation of the PaO2/FiO2 ratio. The procedure was performed by a trained anesthesiologist who was not involved in outcome assessment to maintain blinding.
Outcomes
The primary outcome: VAS score at quiet breathing 30 min after completion of the ICNB.
The secondary outcome: VAS scores at quiet breathing assessed at 6, 12, 18, and 24 h post-intervention; number of rescue analgesic events with flurbiprofen axetil and tramadol within specific time intervals (30 min–6 h, 6–12 h, 12–18 h, 18–24 h); number of patients requiring rescue thoracic paravertebral block (TPVB) within 24 h; diaphragmatic excursion during quiet and deep breathing at 30 min post-intervention; PaO2, SpO2, oxygenation index, and PaCO2 at 30 min post-intervention; incidence of adverse events within 24 h (e.g., hypertension, hypotension, tachycardia, bradycardia, hypoxemia, shortness of breath, allergic reactions, systemic toxicity, pneumothorax, hematoma, bleeding); patient satisfaction with analgesia at 24 h. Hypertension was defined as a systolic blood pressure (SBP) > 140 mmHg or an increase in SBP > 20% from baseline, whichever was higher. Tachycardia was defined as a heart rate (HR) > 100 beats per minute (bpm). Bradycardia was defined as HR < 60 bpm. Hypoxemia was defined as PaO2 < 60 mmHg or SpO2 < 90% while breathing room air. Shortness of breath was defined as the subjective feeling of difficult or uncomfortable breathing, or the inability to get enough air.
The sum of pain intensity difference (SPID) is a widely used metric in clinical trials for evaluating the overall efficacy of an analgesic intervention over a specified period [17]. In our study, sum of pain intensity difference over 0–24 h (SPID0–24h) was calculated based on pain scores at breathing pain scores assessed at 30 min, 6, 12, 18, and 24 h post-intervention across all three groups. The calculation method for SPID0–24h was referenced from established literature [18].
Rescue Analgesia Protocol and Pain Assessment
If a patient's quiet breathing VAS score was ≥ 4 at any time within 24 h post-intervention, a rescue analgesia protocol was initiated: intravenous flurbiprofen axetil (50 mg) was administered first; if flurbiprofen axetil had already been used within 6 h, intravenous tramadol (50 mg) was administered instead (not repeated within 6 h); if the quiet breathing VAS score remained ≥ 4 after sequential administration of these two drugs, analgesic failure was confirmed, and ultrasound-guided TPVB was ultimately performed as rescue analgesia. For patients requiring rescue TPVB because of inadequate analgesia, the last available pre-TPVB VAS score was carried forward for all subsequent time points according to the last observation carried forward principle.
Patient Satisfaction
Patient satisfaction with analgesia was assessed 24 h post-intervention using a survey instrument comprising five questions, each rated on a 5-point Likert scale (1 = strongly disagree; 2 = disagree; 3 = neutral; 4 = agree; 5 = strongly agree) [19]. The questions evaluated the adequacy of attention received for pain treatment, the perceived effectiveness of the pain treatment, the perception of improvement in breathing, the ability to cooperate with lung function exercises, and overall satisfaction with the care provided. A total satisfaction score was calculated by summing the points from all five questions (range, 5 to 25), which was subsequently categorized as excellent (25–21 points), good (20–16 points), fair (15–11 points), or poor (10–5 points).
Statistical Analysis
Based on preliminary data from our institution and pilot study results, with an assumed mean between-group difference of 2 points on the VAS and a standard deviation of 3, a sample size of 45 patients per group was required to achieve 80% power at a two-sided alpha level of 0.025 (adjusted for multiple comparisons). Accounting for a potential 10% dropout rate, a total of 150 patients were recruited.
Statistical analyses were performed using SPSS 26.0. Normality was assessed using the Shapiro-Wilk test. Continuous data are presented as mean ± SD or median (Q1, Q3) and were compared using one-way ANOVA or the Kruskal-Wallis test, as appropriate. Categorical data are presented as n (%) and were compared using the chi-square or Fisher’s exact test. Post hoc pairwise comparisons were performed with Bonferroni correction. A two-sided p < 0.05 was considered statistically significant.

Comments (0)