Findings of SCZ studies
The primary conclusion drawn from the findings of individual omics studies on SCZ is that the pathophysiology of the disorder could be influenced by the effects of environmental factors on various molecular levels, particularly the dopaminergic, glutamatergic, and inflammatory pathways [2].
Regarding multi-omics approaches, researchers have suggested that integrated omics studies on SCZ can be divided into four perspectives: pathogenesis, classification, risk prediction, and precise intervention [2]. These perspectives have been investigated by a variety of integrative omics models, such as genomics–transcriptomics, genomics–epigenomics (a methylome-wide association study), genomics–metabolomics, and genomics–connectomics (i.e., analyzing genomics and the connections within the brain). For instance, regarding the pathogenesis perspective, a transcriptomic imputation analysis, which combines expression quantitative trait locus reference panels with large-scale genotype data, used gene expression predictors to analyze the dorsolateral prefrontal cortex and other brain regions and identified 413 genome-wide significant associations (including 67 nonmajor histocompatibility complex genes, 14 of which were novel) across 13 brain regions and 36 significantly enriched pathways, including pathways involved in hexosaminidase A deficiency and multiple porphyric disorder [8].
In the classification perspective, another transcriptome imputation study offered an analytical approach that used GWAS-predicted gene expression levels and clinical data to classify individuals with SCZ into three subgroups that showed significant differences in treatment response and prognosis [9].
In relation to the risk prediction perspective of SCZ, an integrative analysis of genotype data (from GWASs) and gene expression/regulation data (from functional genomics) embedded the gene regulatory network into a deep-learning model and consequently improved the accuracy of the SCZ prediction model approximately sixfold compared with the conventional single nucleotide polymorphism (SNP)-based polygenic risk scores (PRS) [10].
Regarding the precise intervention perspective, a transcriptome-wide association study used a pharmacogenomic approach, which links genetic variants to antipsychotics response, to compare the differences between the imputed transcriptome from SCZ GWASs and drug-induced gene expression profiles [11]. The study highlighted a novel framework for medication repositioning (i.e., finding new indications for existing medications) and found that repositioning candidates were enriched for multiple antipsychotics. It also identified some potential new medications for SCZ [11].
In human brain tissue, a concurrent transcriptomics, proteomics, and metabolomics study found abnormalities in these molecular layers that were linked to glucoregulatory responses in SCZ [12]. Other researchers have proposed that in individuals with SCZ, elevated glucose demand within the prefrontal cortex is linked to abnormal glucose profiles, which are accompanied by decreased glycolysis and glycogenesis and increased glycogenolysis [13]. So far, transcriptomic analyses have discovered several alternative splicing processes for some of the SCZ risk genes identified by genomic research; interestingly, most of these processes occur in genes that are important for neurodevelopment, neuroplasticity, and cognition [14]. Because miss-splicing events may be involved in SCZ etiology, identifying alternative splicing mechanisms for SCZ risk genes and determining the functional implications of these genes’ altered messenger RNA (mRNA) expression is essential for translating genetic discoveries into novel therapeutic approaches [14]. A transcriptomics-neuroimaging study on transforming growth factor-beta 1 (TGF-β1; an important cytokine for brain development and regenerative processes) found that individuals with SCZ exhibited higher levels of TGF-β1 at the mRNA and protein levels and a significantly thinner cortex in the lateral occipital region than controls; in line with these findings, the individuals with SCZ showed a negative correlation between TGF-β1 levels and visual cognition [15].
In SCZ, the application of genomics-neuroimaging models offers insight into a possible gene-related pathogenesis from the perspective of changes in brain structure and function. One review paper presented results from several magnetic resonance imaging studies that investigated how distinct SNPs in the coding and noncoding regions of various genes affect the functional and structural connectivity and structure of the brain [16]. The authors mentioned that the trans-scale and multi-omics analysis approach that integrates genomics, connectomics, and radiomics is practical for visualizing the connection between functional genetic variations and the neuroanatomical heterogeneity of SCZ [16]. By integrating genomic, transcriptomics, and neuroimaging data from individuals with SCZ and classifying individuals of two SCZ cohorts into broad neural cell-based subtypes, the authors showed that interindividual variations in cell type-specific functions were associated with changes in cortical thickness [17]. Additionally, by applying SCZ risk alleles enriched in cellular functions, they verified the cell-based classification against the genetic variation unique to each patient [17].
A study that combined data from genomics, diffusion tensor imaging, and resting-state functional magnetic resonance imaging showed that higher PRS for SCZ are linked to disrupted white matter integrity and functional connectivity in individuals with SCZ [18]. An integrative analysis of multi-omics data, i.e., of genomics, serum metabolomics, fecal metagenomics, and neuroimaging data, found that the altered metabolome and dysregulated microbiome were associated with neuroactive metabolites, including gamma-aminobutyric acid (GABA), tryptophan, and short-chain fatty acids [19]. It also found that changes in the GABA and tryptophan neurotransmitter pathways were associated with PRS for SCZ and that GABA perhaps plays a more significant role than the other neuroactive metabolites [19]. Another multi-omics investigation on SCZ that used data from genomics, transcriptomics, neuroimaging, and clinical measurements identified 19 distinct genes that were associated with 16 brain areas [20]. Subtype analyses based on gray matter alterations in the target brain region provided a comprehensive understanding of how genetic variations may impact brain architecture and, thus, result in various disorder manifestations [1, 20].
Findings of BD studies
One study used a multi-omics approach to examine the microbiota–gut–brain axis in medication-free individuals with BD [21]. The authors found that alterations in the microbiota were linked to changes in neuroactive metabolites such as pantothenic acid, riboflavin, folic acid, pyridoxine, kynurenic acid, GABA, and short-chain fatty acids. Additionally, in functional imaging scans they found differences in connectivity in relevant areas, such as the hippocampus, amygdala, superior temporal gyrus, and sensorimotor gyrus [21].
The single-cell sequencing technique presents advantages for multi-omics approaches because it can capture cell-type specific characteristics and reduce heterogeneity. One study applied the single-cell disease relevance score method to integrate single-cell resolution RNA sequencing and PRS based on embryonic and fetal brain tissue (i.e., the dorsolateral prefrontal cortex, dorsal pallium, and cortical plate) [22]. The authors analyzed four BD-related omics panels, including brain single-cell RNA-seq data, cell transposase-accessible chromatin using sequencing (ATAC-seq) data, bulk-RNA sequencing data, and GWAS and transcriptome-wide association studies (TWAS) data and found a novel cell cluster expressing adenylate cyclase 1 in the human brain. Moreover, they showed that astrocytes, microglia, and oligodendrocyte precursor cells were significantly associated with BD in different analyses and that the BD-associated genes presented particular characteristics in the various omics assessments [22].
A favorable response to lithium—the first-line treatment in BD—helps to reduce the burden of the condition; however, biomarkers related to this treatment phenotype are lacking. A review of various omics studies in this field found that the target genes related to BD show the involvement of the brain-derived neurotrophic factor gene (BDNF; a protein coding gene involved in neuronal survival, synaptic transmission, and plasticity) in both BD and lithium response [23]. Although methylation patterns in individuals with BD were contradictory and inconclusive, lithium response was partially associated with epigenetic changes through distinct methylation patterns and changes in related enzymes, whereas microRNA alterations remained a promising explanation that still requires further research.
A genomics–transcriptomics study in individuals with BD used data from induced pluripotent stem cell-derived neurons and GWASs on lithium response in responders and nonresponders to obtain candidates related to lithium response phenotype. A total of 1119 candidates were selected, and gene enrichment analysis showed that processes related to the extracellular matrix and focal adhesion were the most relevant [3]. The authors hypothesized that in lithium responders, the extracellular matrix shows defects that impact focal adhesion and are therefore the pathophysiological cause of BD. In these individuals, lithium helps revert the defects and reverse the symptoms of BD, which explains these individuals’ favorable response to the drug [3]. Finally, pharmacometabolomics studies showed that individuals with BD present altered lipid metabolites in the brain and plasma [23].
Findings of MDD studies
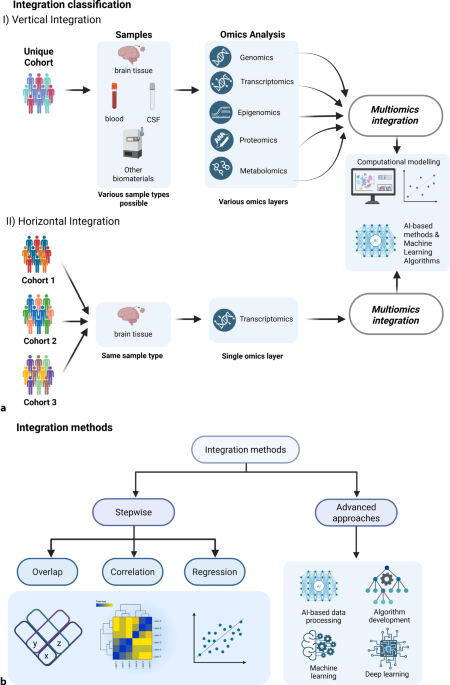
A review article on mood disorders considered eight studies that used multi-omics models in individuals with major depression [24]. In these studies, the data integration strategies utilized included the step-by-step strategy, which detects only strong variance, and the advanced strategy, which makes use of ML/deep learning methods (Fig. 1b).
The step-by-step strategy includes overlapping, correlation, and regression approaches (Fig. 1b). One study used the overlapping approach to compare response to escitalopram in responders, nonresponders, and healthy controls and found 16 differentially methylated CG sites in relevant genes related to neurodevelopmental hippocampal axon pruning and synaptic plasticity [25]. Another study also used this approach to analyze mRNA and DNA methylation in individuals with MDD and controls; it showed that the overlapped hypomethylated and upregulated genes were related to phosphatidylinositol 3‑kinase/protein kinase B (PI3K-Akt), interleukin 17, and axon guidance signaling pathways, whereas the overlapped hypermethylated and downregulated genes were related to the mitogen-activated protein kinase (MAPK), and nuclear factor k-light-chain-enhancer of activated B cells (NF-kB) signaling pathways [26]. The correlation approach was used in a GWAS in twins discordant for MDD, and the study found overrepresentation of differentially methylated genes in the individuals with MDD [27]. Another study used the regression approach to understand the mRNA, microRNA, and clinical correlates of treatment-worsening suicidal ideation and found that a logistic model including miR-5695, STMN1 mRNA, and the Montgomery–Asberg Depression Rating Scale score at baseline predicted this outcome [28].
The advanced strategy was used by two studies. The first integrated data from GWAS and data on expression quantitative trait locus, RNA-seq in the dorsolateral prefrontal cortex, and chromatin conformation (Hi-C) from large cohorts of individuals with MDD and found two implicated genes related to (i) synapse formation and differentiation (LRFN5) and (ii) synaptic plasticity (DCC) [29]. The second study used differentially methylated CpG sites to develop a random forest classifier to detect suicidal behavior and showed an accuracy of 92.6% [30].
The diagnosis of depression remains a challenge because of the lack of valid biomarkers. Aiming to identify such biomarkers, a scoping review evaluated studies that applied ML models to omics data for clustering individuals with depression [4]. The article included 15 studies that used various omics types, such as genomics, epigenomics, transcriptomics, and microbiomics, to evaluate samples from blood, brain, and stool. Among these studies, only one integrated multi-omics data by using a random forest classifier to accurately predict suicidal behavior (see above [30]). Another study used ML analysis of microRNA expression data to differentiate between medication-free individuals with MDD and controls and showed high accuracy of the approach (AUC = 0.97) [31]. Moreover, the authors were able to separate individuals with mild MDD from those with moderate or severe MDD by using unsupervised ML in two clusters of patients (AUCs of 70 and 76).
Antidepressant treatment represents another challenge because of the high interindividual heterogeneity in dose response and tolerance. In the case of treatment-resistant depression, multi-omics approaches have revealed an association with alterations related to i) the immune system, inflammatory pathways, and the hypothalamic–pituitary–adrenal (HPA) axis; ii) neuroplasticity; iii) calcium signaling; iv) neurotransmitters; and v) other factors, such as the cytoskeleton, blood coagulation, and apoptosis/autophagy. These findings were supported by genomics, transcriptomics, and metabolomics data, among others [32].



Comments (0)